Quesada-Romero Luisa, Mena-Ulecia Karel, Tiznado William, Caballero Julio
Centro de Bioinformática y Simulación Molecular, Facultad de Ingeniería, Universidad de Talca, Talca, Chile.
Departamento de Ciencias Químicas, Facultad de Ciencias Exactas, Universidad Andres Bello, Santiago de Chile, Chile.
PLoS One. 2014 Jul 10;9(7):e102212. doi: 10.1371/journal.pone.0102212. eCollection 2014.
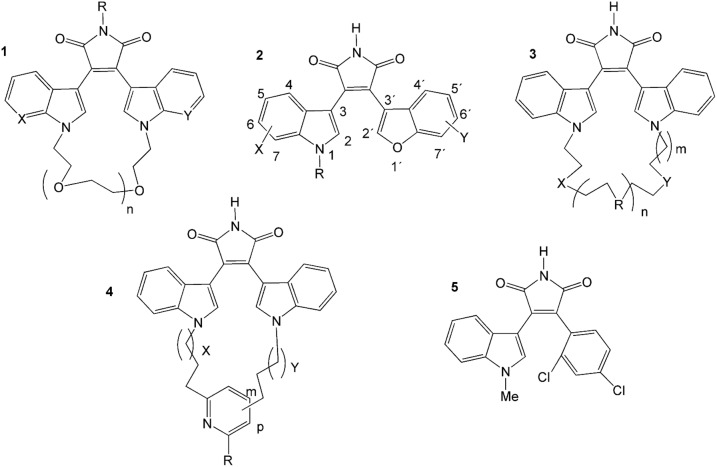
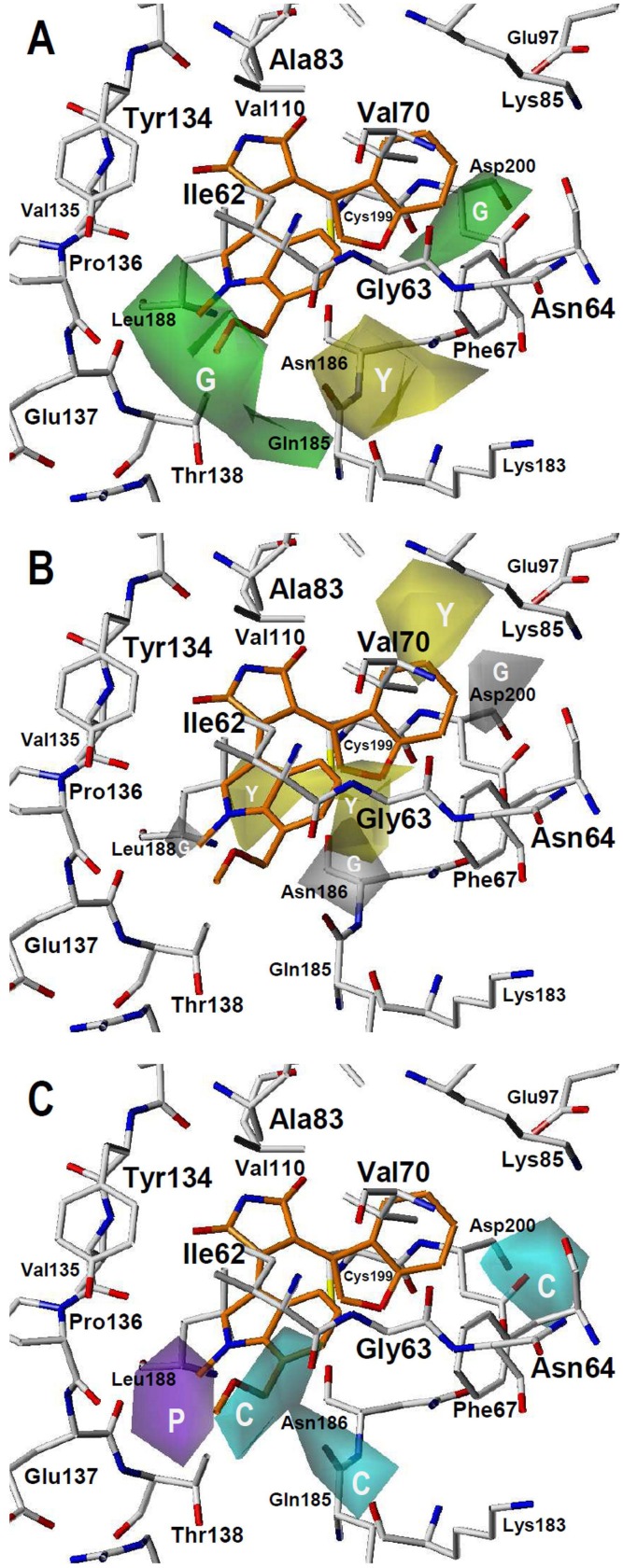
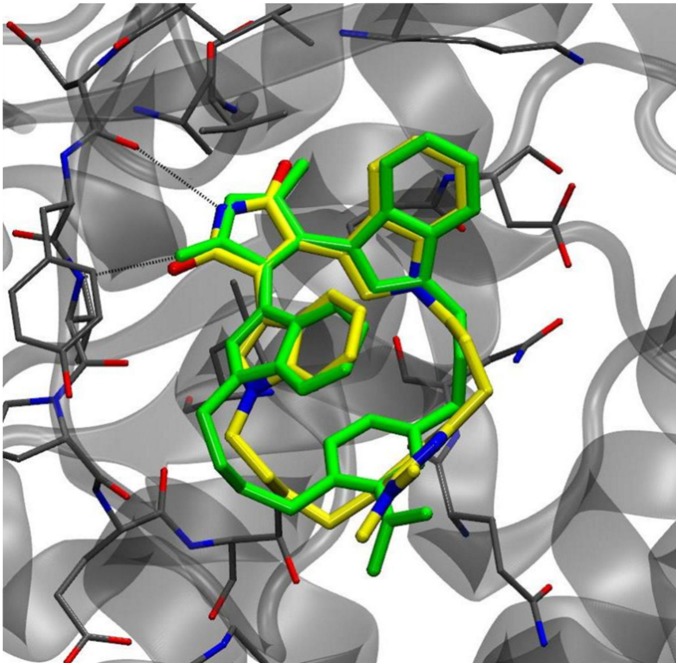
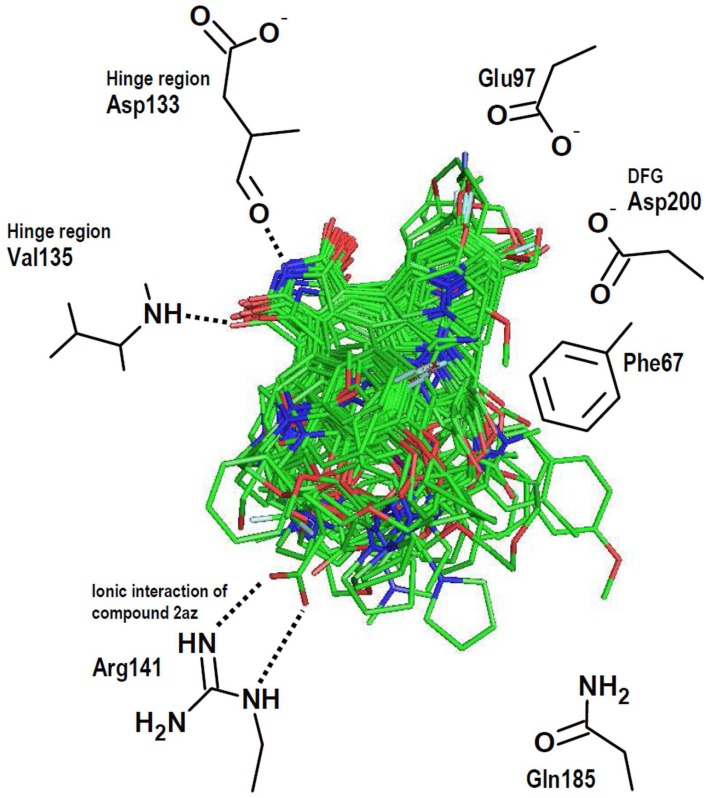
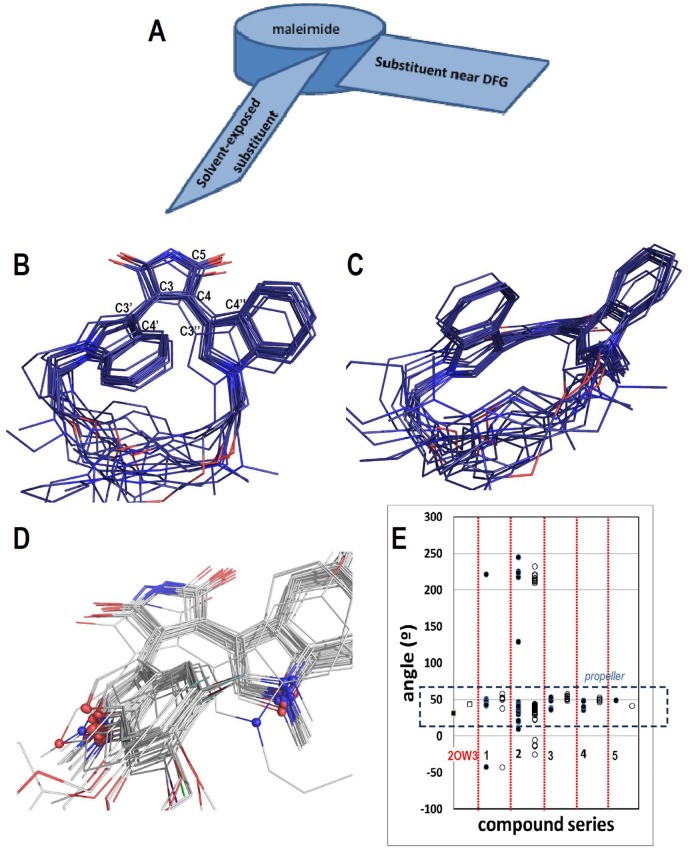
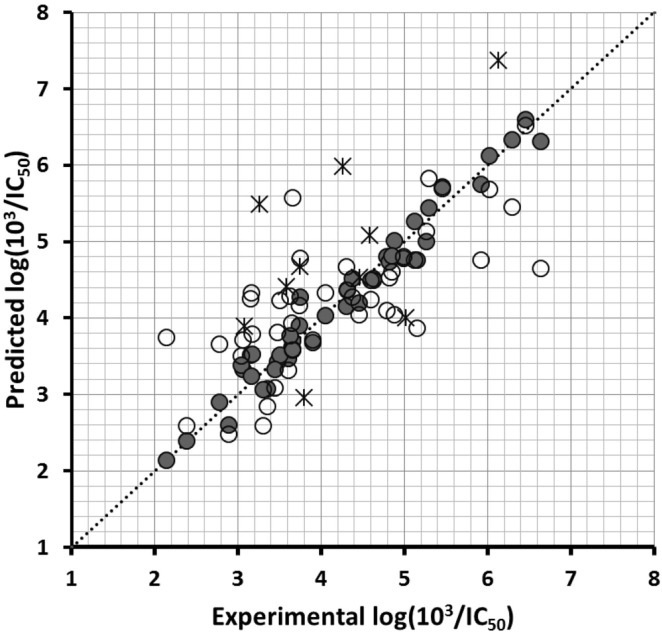
Many protein kinase (PK) inhibitors have been reported in recent years, but only a few have been approved for clinical use. The understanding of the available molecular information using computational tools is an alternative to contribute to this process. With this in mind, we studied the binding modes of 77 maleimide derivates inside the PK glycogen synthase kinase 3 beta (GSK3β) using docking experiments. We found that the orientations that these compounds adopt inside GSK3β binding site prioritize the formation of hydrogen bond (HB) interactions between the maleimide group and the residues at the hinge region (residues Val135 and Asp133), and adopt propeller-like conformations (where the maleimide is the propeller axis and the heterocyclic substituents are two slanted blades). In addition, quantitative structure-activity relationship (QSAR) models using CoMSIA methodology were constructed to explain the trend of the GSK3β inhibitory activities for the studied compounds. We found a model to explain the structure-activity relationship of non-cyclic maleimide (NCM) derivatives (54 compounds). The best CoMSIA model (training set included 44 compounds) included steric, hydrophobic, and HB donor fields and had a good Q(2) value of 0.539. It also predicted adequately the most active compounds contained in the test set. Furthermore, the analysis of the plots of the steric CoMSIA field describes the elements involved in the differential potency of the inhibitors that can be considered for the selection of suitable inhibitors.
近年来已报道了许多蛋白激酶(PK)抑制剂,但只有少数被批准用于临床。利用计算工具了解可用的分子信息是有助于这一过程的一种方法。考虑到这一点,我们通过对接实验研究了77种马来酰亚胺衍生物在PK糖原合酶激酶3β(GSK3β)内的结合模式。我们发现这些化合物在GSK3β结合位点内所采取的取向优先考虑马来酰亚胺基团与铰链区残基(Val135和Asp133残基)之间形成氢键(HB)相互作用,并采取螺旋桨状构象(其中马来酰亚胺为螺旋桨轴,杂环取代基为两个倾斜叶片)。此外,构建了使用CoMSIA方法的定量构效关系(QSAR)模型来解释所研究化合物的GSK3β抑制活性趋势。我们发现了一个解释非环状马来酰亚胺(NCM)衍生物(54种化合物)构效关系的模型。最佳的CoMSIA模型(训练集包括44种化合物)包括空间、疏水和HB供体场,具有良好的Q(2)值0.539。它还充分预测了测试集中最具活性化合物。此外,对空间CoMSIA场图的分析描述了抑制剂差异效力中涉及的元素,这些元素可用于选择合适的抑制剂。