Centro de Bioinformática y Simulación Molecular (CBSM), Universidad de Talca, Talca 3460000, Chile.
Int J Mol Sci. 2018 Sep 28;19(10):2956. doi: 10.3390/ijms19102956.
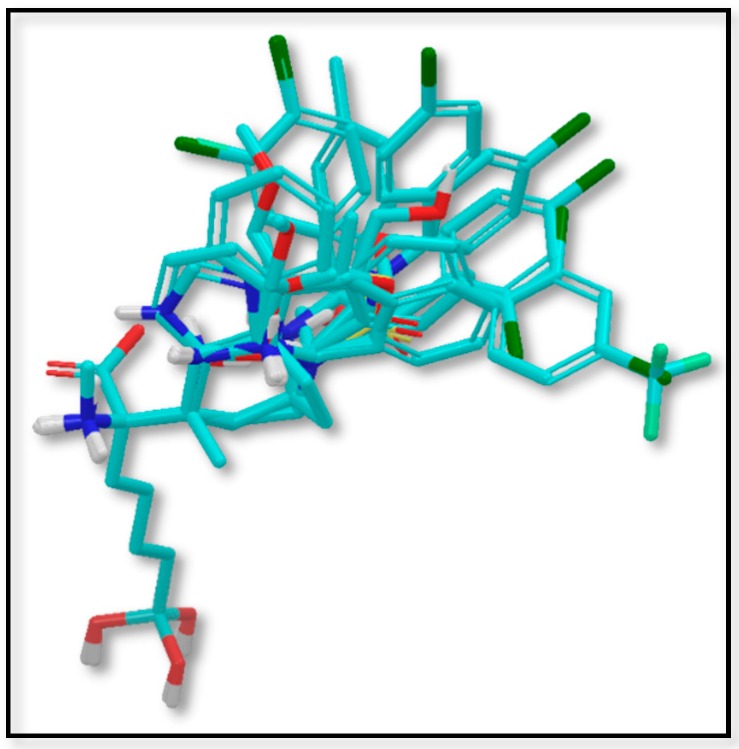
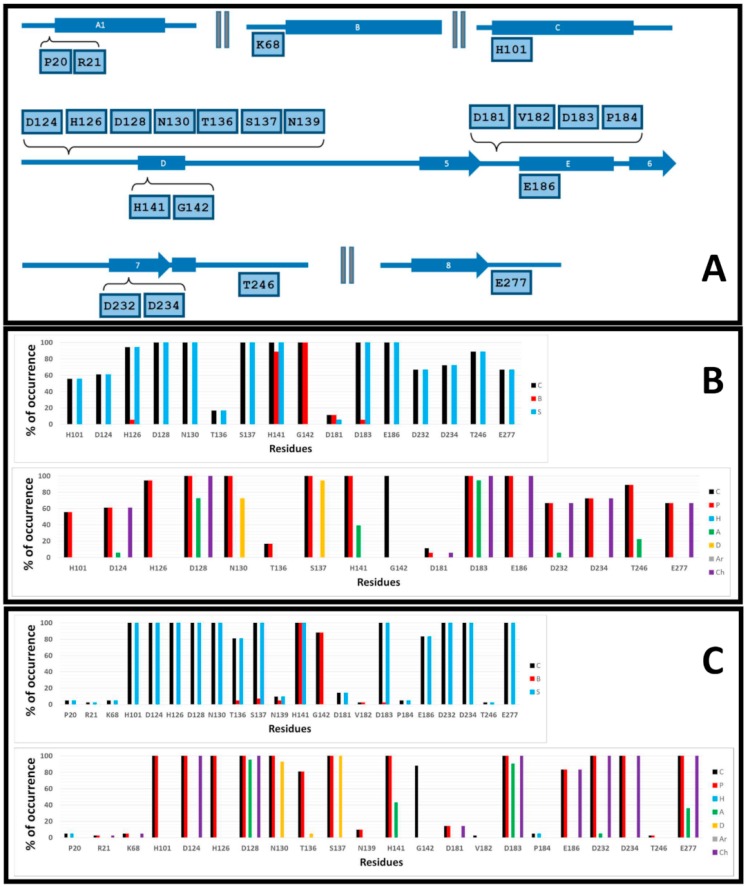
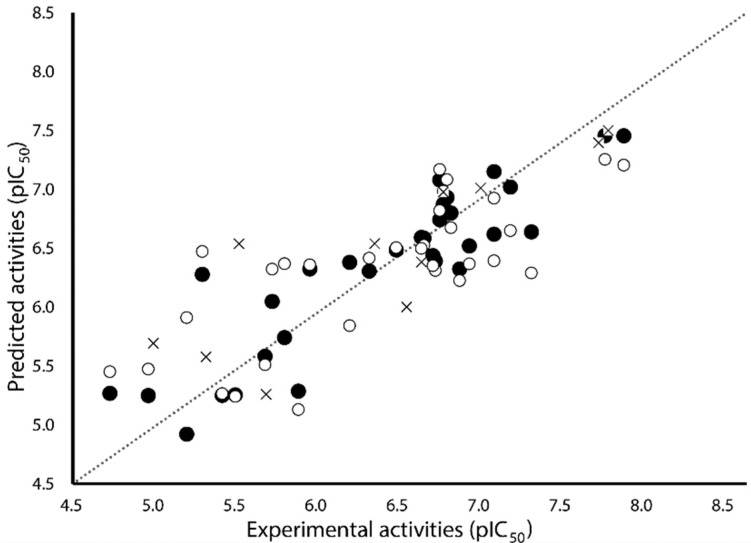
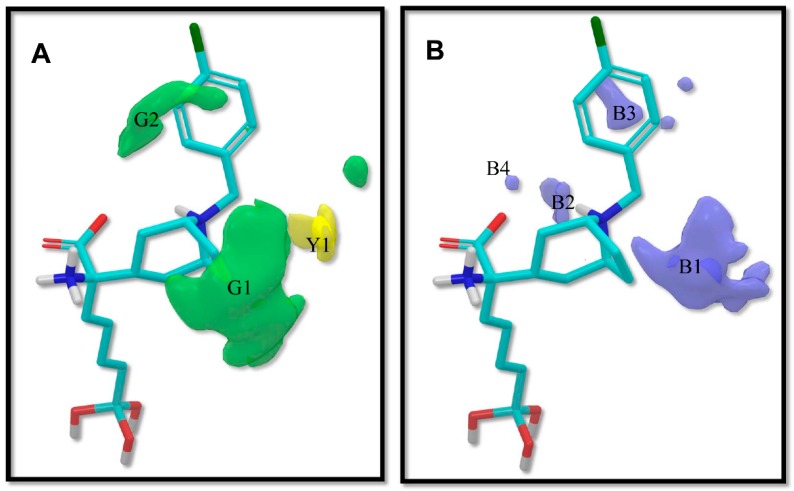
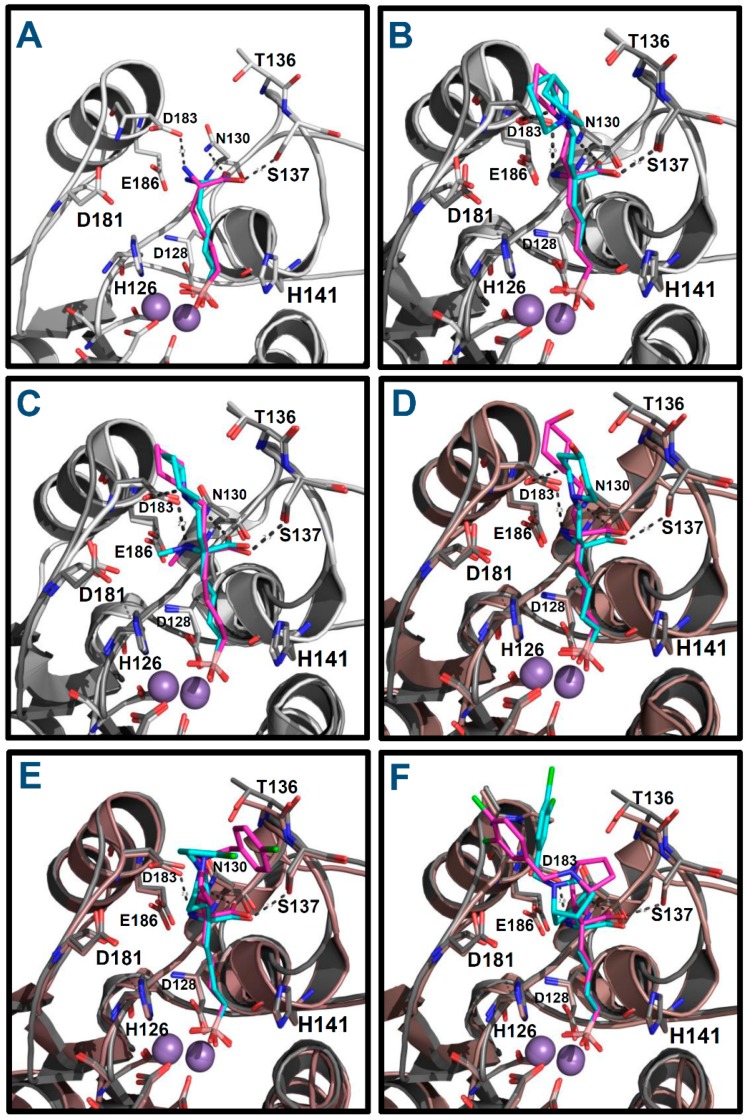
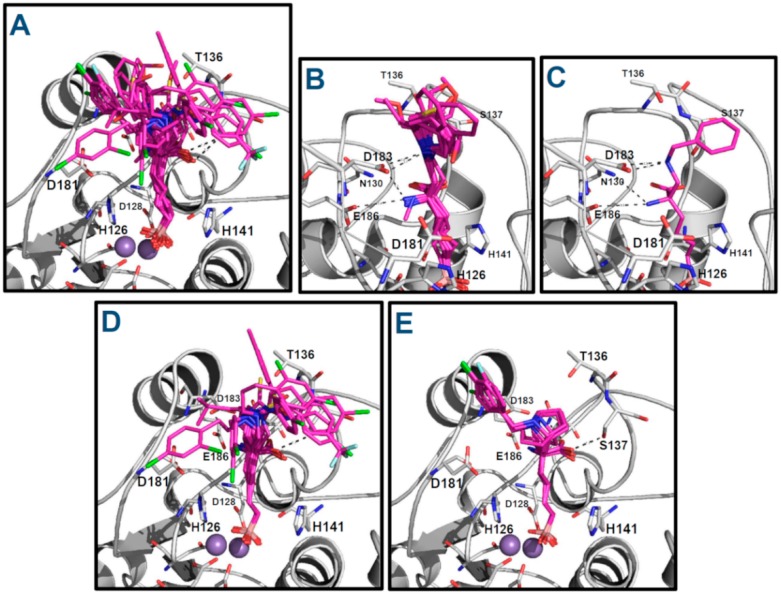
Human arginase I (hARGI) is an important enzyme involved in the urea cycle; its overexpression has been associated to cardiovascular and cerebrovascular diseases. In the last years, several congeneric sets of hARGI inhibitors have been reported with possible beneficial roles for the cardiovascular system. At the same time, crystallographic data have been reported including hARGI⁻inhibitor complexes, which can be considered for the design of novel inhibitors. In this work, the structure⁻activity relationship (SAR) of Cα substituted 2(S)-amino-6-boronohexanoic acid (ABH) derivatives as hARGI inhibitors was studied by using a three-dimensional quantitative structure⁻activity relationships (3D-QSAR) method. The predictivity of the obtained 3D-QSAR model was demonstrated by using internal and external validation experiments. The best model revealed that the differential hARGI inhibitory activities of the derivatives can be described by using steric and electrostatic fields; the local effects of these fields in the activity are presented. In addition, binding modes of the above-mentioned compounds inside the hARGI binding site were obtained by using molecular docking. It was found that derivatives adopted the same orientation reported for within the hARGI active site, with the substituents at Cα exposed to the solvent with interactions with residues at the entrance of the binding site. The hARGI residues involved in chemical interactions with inhibitors were identified by using an interaction fingerprints (IFPs) analysis.
人精氨酸酶 I(hARGI)是参与尿素循环的重要酶;其过表达与心血管和脑血管疾病有关。在过去的几年中,已经报道了几类同源的 hARGI 抑制剂,它们可能对心血管系统有有益的作用。同时,也报道了晶体学数据,包括 hARGI-抑制剂复合物,可以考虑用于设计新型抑制剂。在这项工作中,通过使用三维定量构效关系(3D-QSAR)方法研究了 Cα 取代的 2(S)-氨基-6-硼代己酸(ABH)衍生物作为 hARGI 抑制剂的结构-活性关系(SAR)。通过内部和外部验证实验证明了获得的 3D-QSAR 模型的预测能力。最佳模型表明,可以使用立体和静电场来描述衍生物的 hARGI 抑制活性的差异;呈现了这些场在活性中的局部影响。此外,通过分子对接获得了上述化合物在 hARGI 结合位点内的结合模式。发现衍生物在 hARGI 活性位点内采用了与[1]报告的相同的取向,Cα 上的取代基暴露在溶剂中,与结合位点入口处的残基相互作用。通过相互作用指纹(IFPs)分析确定了与抑制剂发生化学相互作用的 hARGI 残基。