Division of Biochemistry, Netherlands Cancer Institute , Plesmanlaan 121, 1066 CX Amsterdam, The Netherlands.
Structural Studies Division, MRC Laboratory of Molecular Biology , Francis Crick Avenue, Cambridge CB2 0HQ, England.
IUCrJ. 2014 May 30;1(Pt 4):213-20. doi: 10.1107/S2052252514009324. eCollection 2014 Jul 1.
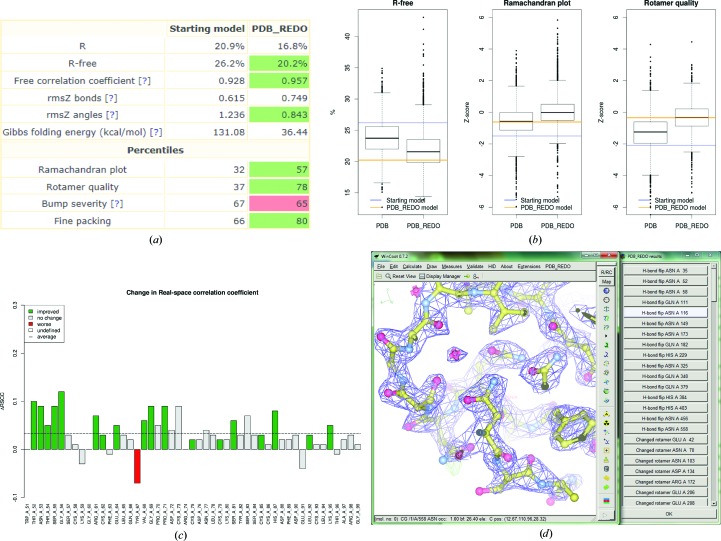
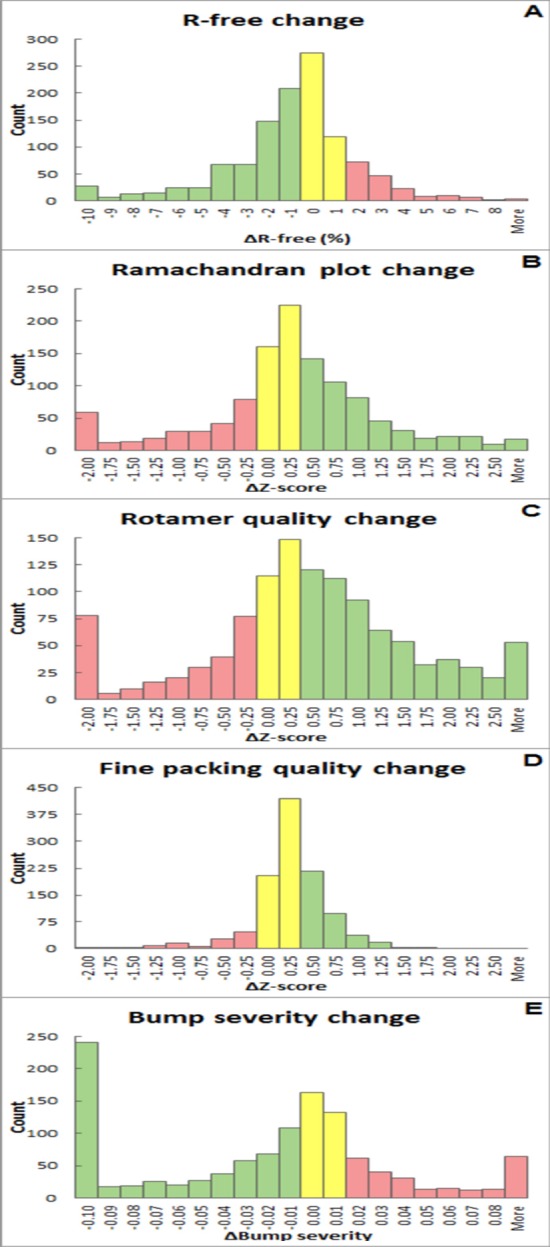
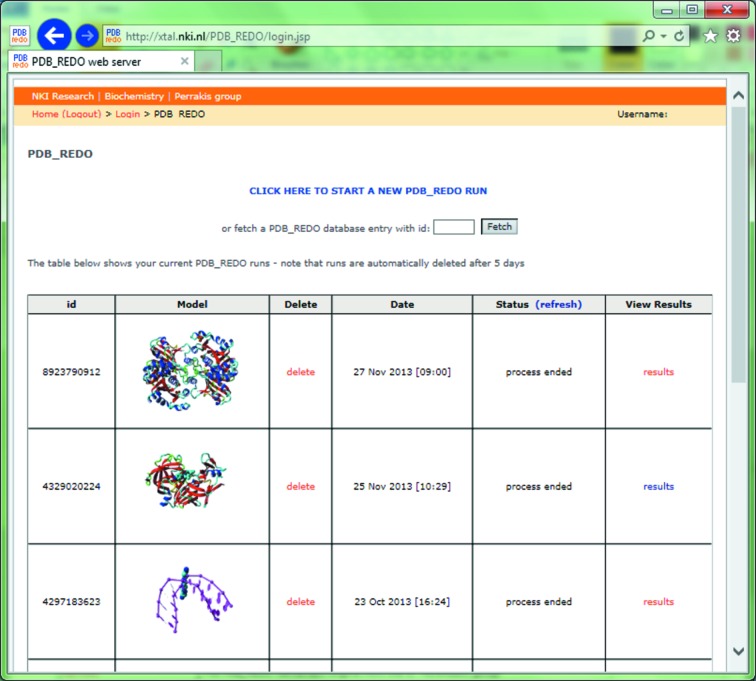
The refinement and validation of a crystallographic structure model is the last step before the coordinates and the associated data are submitted to the Protein Data Bank (PDB). The success of the refinement procedure is typically assessed by validating the models against geometrical criteria and the diffraction data, and is an important step in ensuring the quality of the PDB public archive [Read et al. (2011 ▶), Structure, 19, 1395-1412]. The PDB_REDO procedure aims for 'constructive validation', aspiring to consistent and optimal refinement parameterization and pro-active model rebuilding, not only correcting errors but striving for optimal interpretation of the electron density. A web server for PDB_REDO has been implemented, allowing thorough, consistent and fully automated optimization of the refinement procedure in REFMAC and partial model rebuilding. The goal of the web server is to help practicing crystallo-graphers to improve their model prior to submission to the PDB. For this, additional steps were implemented in the PDB_REDO pipeline, both in the refinement procedure, e.g. testing of resolution limits and k-fold cross-validation for small test sets, and as new validation criteria, e.g. the density-fit metrics implemented in EDSTATS and ligand validation as implemented in YASARA. Innovative ways to present the refinement and validation results to the user are also described, which together with auto-generated Coot scripts can guide users to subsequent model inspection and improvement. It is demonstrated that using the server can lead to substantial improvement of structure models before they are submitted to the PDB.
晶体结构模型的精修和验证是将坐标和相关数据提交给蛋白质数据库 (PDB) 之前的最后一步。精修过程的成功通常通过根据几何标准和衍射数据验证模型来评估,这是确保 PDB 公共档案质量的重要步骤[Read 等人,2011▶,结构,19,1395-1412]。PDB_REDO 程序旨在进行“建设性验证”,力求实现一致和最佳的精修参数化和积极的模型重建,不仅纠正错误,而且努力优化电子密度的解释。已经实现了用于 PDB_REDO 的 Web 服务器,允许在 REFMAC 中彻底、一致和全自动优化精修程序,并进行部分模型重建。Web 服务器的目标是帮助晶体学家在将模型提交给 PDB 之前改进模型。为此,在 PDB_REDO 管道中实现了额外的步骤,包括精修过程中的步骤,例如测试分辨率限制和小测试集的 k 折交叉验证,以及新的验证标准,例如在 EDSTATS 中实现的密度拟合指标和在 YASARA 中实现的配体验证。还描述了向用户展示精修和验证结果的创新方法,这些方法与自动生成的 Coot 脚本一起可以指导用户进行后续的模型检查和改进。结果表明,使用该服务器可以在将结构模型提交给 PDB 之前显著提高模型质量。