van Beusekom Bart, Joosten Krista, Hekkelman Maarten L, Joosten Robbie P, Perrakis Anastassis
Department of Biochemistry, The Netherlands Cancer Institute, Plesmanlaan 121, Amsterdam 1066CX, The Netherlands.
IUCrJ. 2018 Aug 8;5(Pt 5):585-594. doi: 10.1107/S2052252518010552. eCollection 2018 Sep 1.
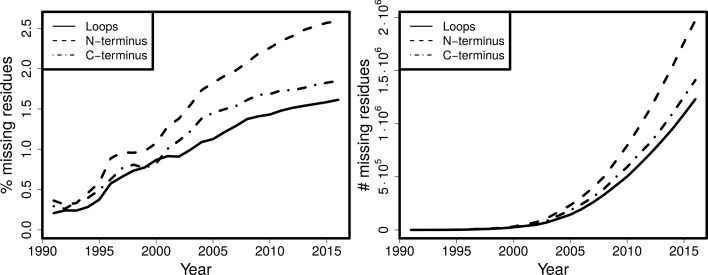
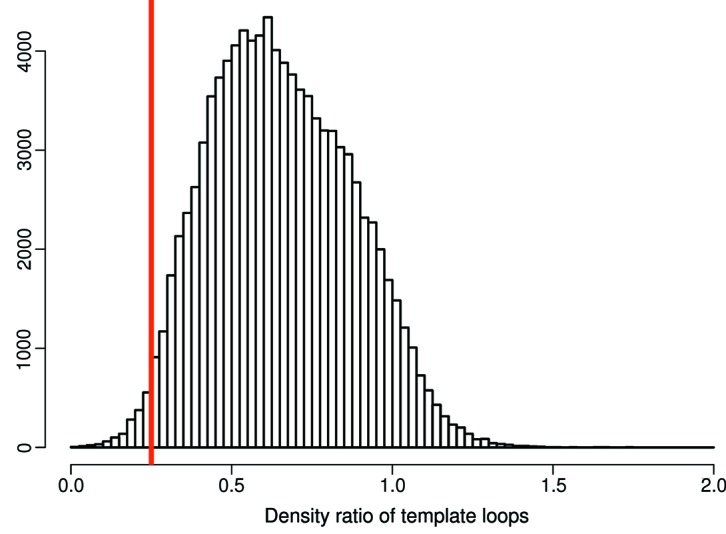
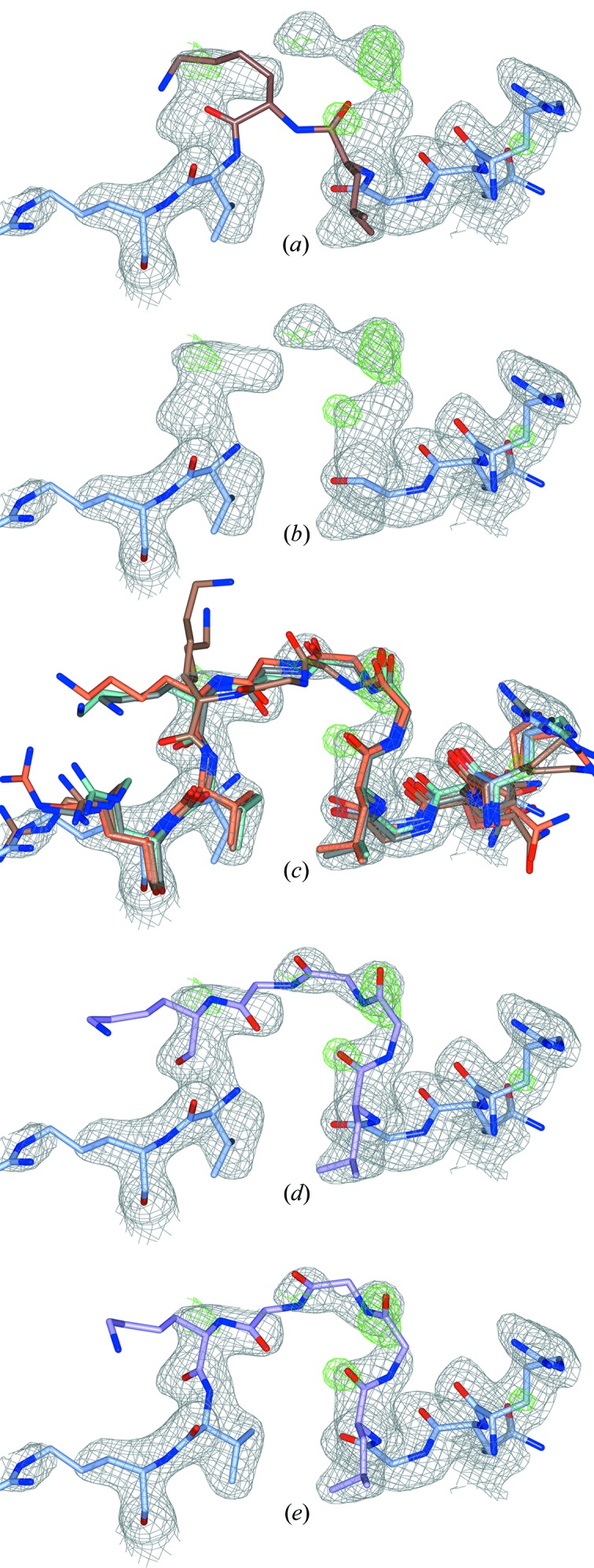
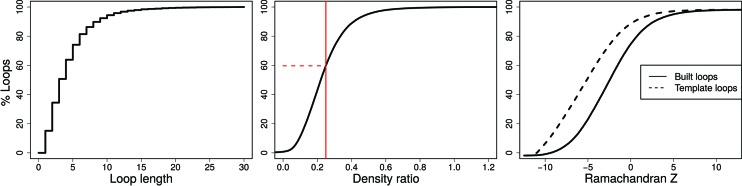
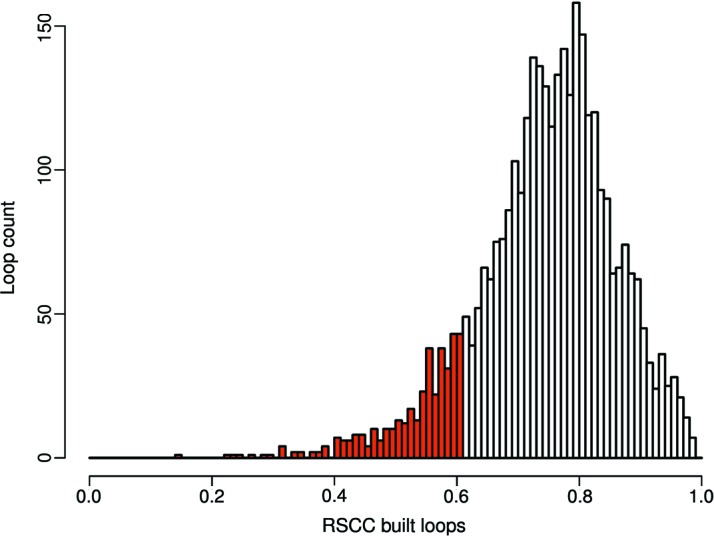
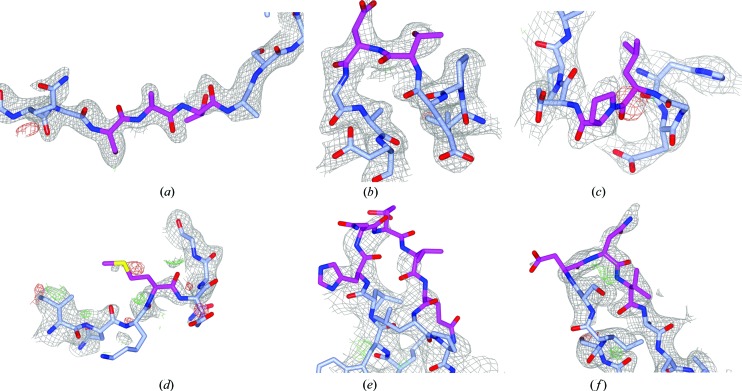
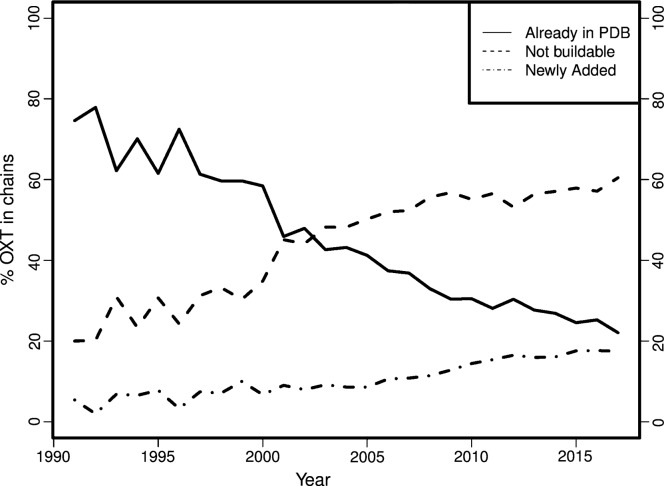
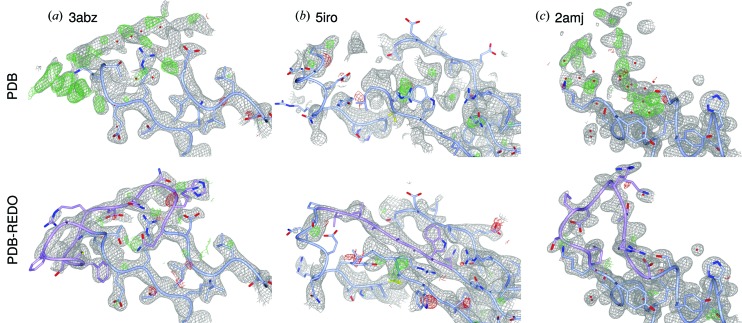
Inherent protein flexibility, poor or low-resolution diffraction data or poorly defined electron-density maps often inhibit the building of complete structural models during X-ray structure determination. However, recent advances in crystallographic refinement and model building often allow completion of previously missing parts. This paper presents algorithms that identify regions missing in a certain model but present in homologous structures in the Protein Data Bank (PDB), and 'graft' these regions of interest. These new regions are refined and validated in a fully automated procedure. Including these developments in the pipeline has enabled the building of 24 962 missing loops in the PDB. The models and the automated procedures are publicly available through the PDB-REDO databank and webserver. More complete protein structure models enable a higher quality public archive but also a better understanding of protein function, better comparison between homologous structures and more complete data mining in structural bioinformatics projects.
内在蛋白质灵活性、较差或低分辨率的衍射数据或定义不明确的电子密度图,在X射线结构测定过程中常常会阻碍完整结构模型的构建。然而,晶体学精修和模型构建方面的最新进展常常能使之前缺失的部分得以补齐。本文介绍了一些算法,这些算法能识别某个模型中缺失但存在于蛋白质数据库(PDB)中同源结构里的区域,并“移植”这些感兴趣的区域。这些新区域会通过一个完全自动化的流程进行精修和验证。将这些进展纳入流程已使得在PDB中构建了24962个缺失的环。这些模型和自动化流程可通过PDB-REDO数据库和网络服务器公开获取。更完整的蛋白质结构模型不仅能形成更高质量的公共存档,还能更好地理解蛋白质功能、更准确地比较同源结构以及在结构生物信息学项目中进行更全面的数据挖掘。