Wild Edward J, Tabrizi Sarah J
Department of Neurodegenerative Disease, UCL Institute of Neurology, National Hospital for Neurology & Neurosurgery, Queen Square, London, WC1N 3BG, UK.
Mov Disord. 2014 Sep 15;29(11):1434-45. doi: 10.1002/mds.26007. Epub 2014 Aug 25.
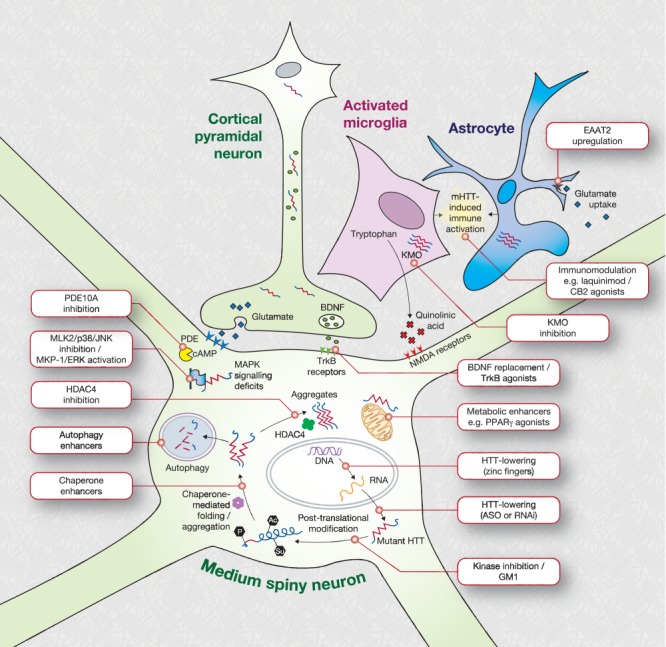
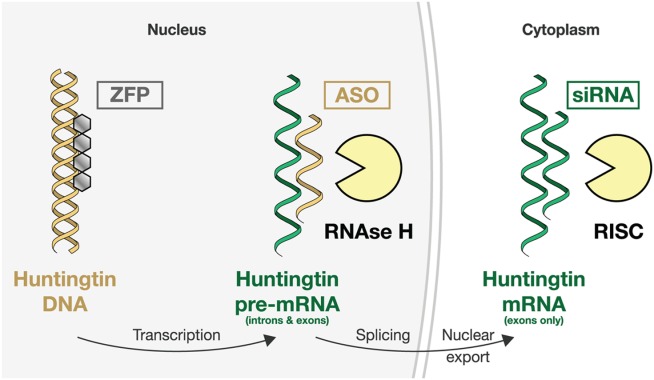
The known genetic cause of Huntington's disease (HD) has fueled considerable progress in understanding its pathobiology and the development of therapeutic approaches aimed at correcting specific changes linked to the causative mutation. Among the most promising is reducing expression of mutant huntingtin protein (mHTT) with RNA interference or antisense oligonucleotides; human trials are now being planned. Zinc-finger transcriptional repression is another innovative method to reduce mHTT expression. Modulation of mHTT phosphorylation, chaperone upregulation, and autophagy enhancement represent attempts to alter cellular homeostasis to favor removal of mHTT. Inhibition of histone deacetylases (HDACs) remains of interest; recent work affirms HDAC4 as a target but questions the assumed centrality of its catalytic activity in HD. Phosphodiesterase inhibition, aimed at restoring synaptic function, has progressed rapidly to human trials. Deranged cellular signaling provides several tractable targets, but specificity and complexity are challenges. Restoring neurotrophic support in HD remains a key potential therapeutic approach. with several approaches being pursued, including brain-derived neurotrophic factor (BDNF) mimesis through tyrosine receptor kinase B (TrkB) agonism and monoclonal antibodies. An increasing understanding of the role of glial cells in HD has led to several new therapeutic avenues, including kynurenine monooxygenase inhibition, immunomodulation by laquinimod, CB2 agonism, and others. The complex metabolic derangements in HD remain under study, but no clear therapeutic strategy has yet emerged. We conclude that many exciting therapeutics are progressing through the development pipeline, and combining a better understanding of HD biology in human patients, with concerted medicinal chemistry efforts, will be crucial for bringing about an era of effective therapies.
亨廷顿舞蹈病(HD)已知的遗传病因推动了在理解其病理生物学以及开发旨在纠正与致病突变相关的特定变化的治疗方法方面取得了相当大的进展。其中最有前景的方法之一是使用RNA干扰或反义寡核苷酸降低突变型亨廷顿蛋白(mHTT)的表达;目前正在计划进行人体试验。锌指转录抑制是另一种降低mHTT表达的创新方法。调节mHTT磷酸化、伴侣蛋白上调和自噬增强代表了改变细胞内稳态以促进mHTT清除的尝试。抑制组蛋白脱乙酰酶(HDACs)仍然受到关注;最近的研究证实HDAC4是一个靶点,但对其催化活性在HD中所假定的核心地位提出了质疑。旨在恢复突触功能的磷酸二酯酶抑制作用已迅速进入人体试验阶段。紊乱的细胞信号传导提供了几个易于处理的靶点,但特异性和复杂性是挑战。在HD中恢复神经营养支持仍然是一种关键的潜在治疗方法,正在探索多种方法,包括通过酪氨酸受体激酶B(TrkB)激动作用和单克隆抗体模拟脑源性神经营养因子(BDNF)。对神经胶质细胞在HD中的作用的日益了解带来了几种新的治疗途径,包括抑制犬尿氨酸单加氧酶、拉喹莫德的免疫调节、CB2激动作用等。HD中复杂的代谢紊乱仍在研究中,但尚未出现明确的治疗策略。我们得出结论,许多令人兴奋的治疗方法正在研发过程中,将对人类患者HD生物学的更好理解与协同的药物化学努力相结合,对于实现有效治疗的时代至关重要。