Martin Eric, Carlson Jonathan M, Le Anh Q, Chopera Denis R, McGovern Rachel, Rahman Manal A, Ng Carmond, Jessen Heiko, Kelleher Anthony D, Markowitz Martin, Allen Todd M, Milloy M-J, Carrington Mary, Wainberg Mark A, Brumme Zabrina L
Retrovirology. 2014 Aug 29;11:64. doi: 10.1186/s12977-014-0064-1.
The reproducible nature of HIV-1 escape from HLA-restricted CD8+ T-cell responses allows the identification of HLA-associated viral polymorphisms "at the population level" - that is, via analysis of cross-sectional, linked HLA/HIV-1 genotypes by statistical association. However, elucidating their timing of selection traditionally requires detailed longitudinal studies, which are challenging to undertake on a large scale. We investigate whether the extent and relative timecourse of immune-driven HIV adaptation can be inferred via comparative cross-sectional analysis of independent early and chronic infection cohorts.
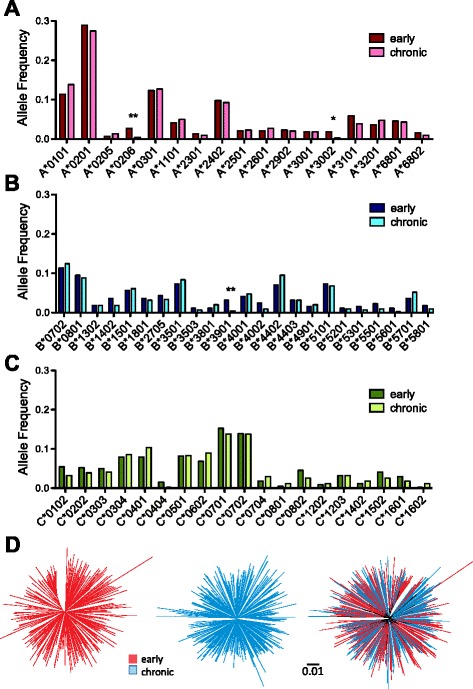
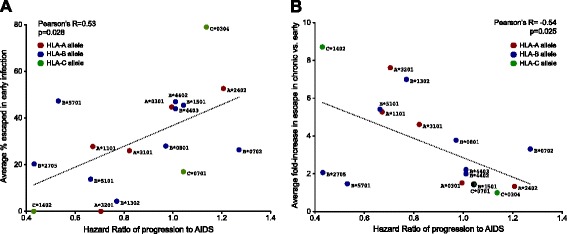
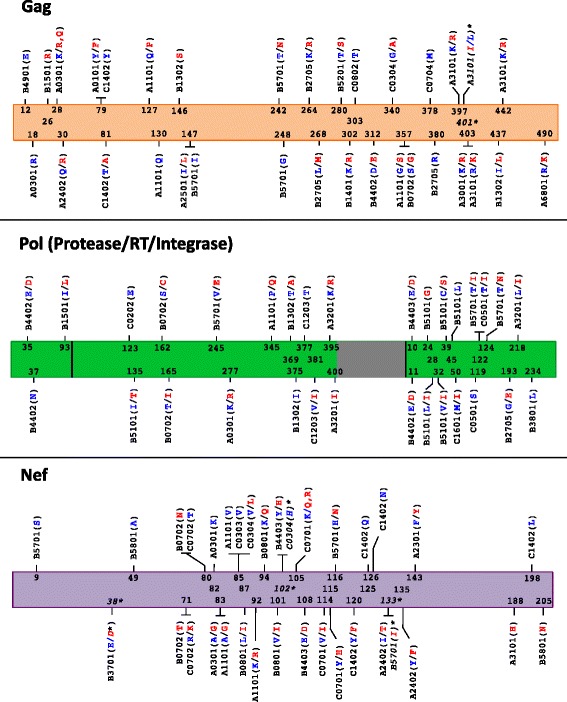
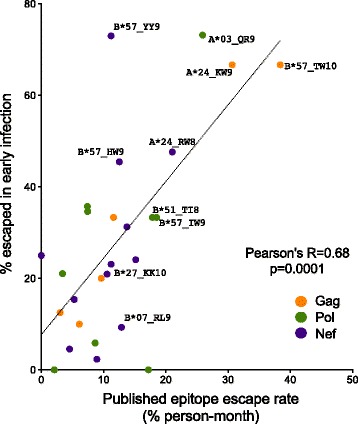
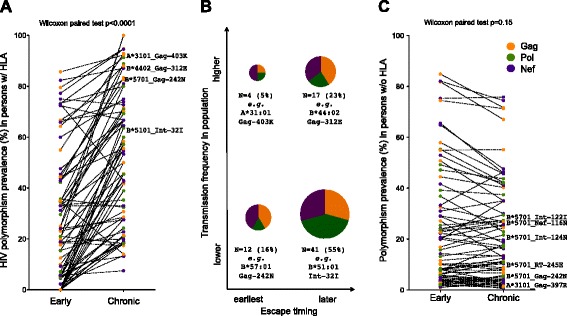
Similarly-powered datasets of linked HLA/HIV-1 genotypes from individuals with early (median < 3 months) and chronic untreated HIV-1 subtype B infection, matched for size (N > 200/dataset), HLA class I and HIV-1 Gag/Pol/Nef diversity, were established. These datasets were first used to define a list of 162 known HLA-associated polymorphisms detectable at the population level in cohorts of the present size and host/viral genetic composition. Of these 162 known HLA-associated polymorphisms, 15% (occurring at 14 Gag, Pol and Nef codons) were already detectable via statistical association in the early infection dataset at p ≤ 0.01 (q < 0.2) - identifying them as the most consistently rapidly escaping sites in HIV-1. Among these were known rapidly-escaping sites (e.g. B57-Gag-T242N) and others not previously appreciated to be reproducibly rapidly selected (e.g. A31:01-associated adaptations at Gag codons 397, 401 and 403). Escape prevalence in early infection correlated strongly with first-year escape rates (Pearson's R = 0.68, p = 0.0001), supporting cross-sectional parameters as reliable indicators of longitudinally-derived measures. Comparative analysis of early and chronic datasets revealed that, on average, the prevalence of HLA-associated polymorphisms more than doubles between these two infection stages in persons harboring the relevant HLA (p < 0.0001, consistent with frequent and reproducible escape), but remains relatively stable in persons lacking the HLA (p = 0.15, consistent with slow reversion). Published HLA-specific Hazard Ratios for progression to AIDS correlated positively with average escape prevalence in early infection (Pearson's R = 0.53, p = 0.028), consistent with high early within-host HIV-1 adaptation (via rapid escape and/or frequent polymorphism transmission) as a correlate of progression.
Cross-sectional host/viral genotype datasets represent an underutilized resource to identify reproducible early pathways of HIV-1 adaptation and identify correlates of protective immunity.
HIV-1逃避HLA限制的CD8+T细胞反应具有可重复性,这使得在“群体水平”上识别HLA相关的病毒多态性成为可能——也就是说,通过统计关联分析横断面的、连锁的HLA/HIV-1基因型。然而,传统上阐明它们的选择时机需要详细的纵向研究,而大规模开展此类研究具有挑战性。我们研究是否可以通过对独立的早期和慢性感染队列进行比较横断面分析,推断免疫驱动的HIV适应性变化的程度和相对时间进程。
建立了来自早期(中位数<3个月)和慢性未治疗的HIV-1 B亚型感染个体的连锁HLA/HIV-1基因型数据集,这些数据集规模相当(每个数据集N>200),并在HLA I类和HIV-1 Gag/Pol/Nef多样性方面进行了匹配。这些数据集首先用于确定在当前规模的队列以及宿主/病毒基因组成中可在群体水平检测到的162个已知HLA相关多态性列表。在这162个已知的HLA相关多态性中,15%(发生在14个Gag、Pol和Nef密码子处)在早期感染数据集中通过统计关联已经可以检测到(p≤0.01,q<0.2)——将它们确定为HIV-1中最一致快速逃逸的位点。其中包括已知的快速逃逸位点(如B57-Gag-T242N)以及其他以前未被认识到可重复性快速选择的位点(如A31:01相关的Gag密码子397、401和403处的适应性变化)。早期感染中的逃逸流行率与第一年的逃逸率密切相关(Pearson相关系数R=0.68,p=0.0001),支持横断面参数作为纵向测量的可靠指标。早期和慢性数据集的比较分析表明,平均而言,在携带相关HLA的人群中,这两个感染阶段之间HLA相关多态性的流行率增加了一倍多(p<0.0001,与频繁且可重复的逃逸一致),但在缺乏该HLA的人群中保持相对稳定(p=0.15,与缓慢回复一致)。已发表的关于进展为艾滋病的HLA特异性风险比与早期感染中的平均逃逸流行率呈正相关(Pearson相关系数R=0.53,p=0.028),这与宿主内早期HIV-1的高适应性(通过快速逃逸和/或频繁的多态性传播)作为疾病进展的一个相关因素一致。
横断面宿主/病毒基因型数据集是一种未充分利用的资源,可用于识别HIV-1适应性变化的可重复早期途径,并确定保护性免疫的相关因素。