Janes Kali, Esposito Emanuela, Doyle Timothy, Cuzzocrea Salvatore, Tosh Dillip K, Jacobson Kenneth A, Salvemini Daniela
Department of Pharmacological and Physiological Science, Saint Louis University School of Medicine, 1402 South Grand Blvd, St Louis, MO 63104, USA Department of Clinical and Experimental Medicine and Pharmacology, University of Messina, Messina 98122, Italy Molecular Recognition Section, Laboratory of Bioorganic Chemistry, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, MD 20892-0810, USA.
Pain. 2014 Dec;155(12):2560-2567. doi: 10.1016/j.pain.2014.09.016. Epub 2014 Sep 19.
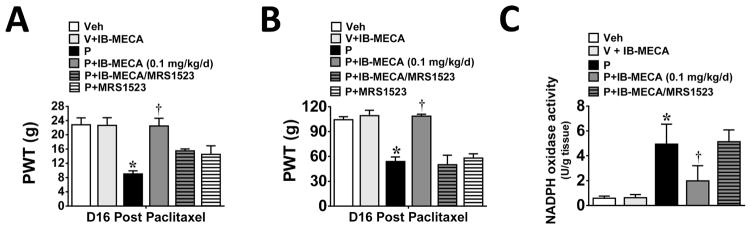
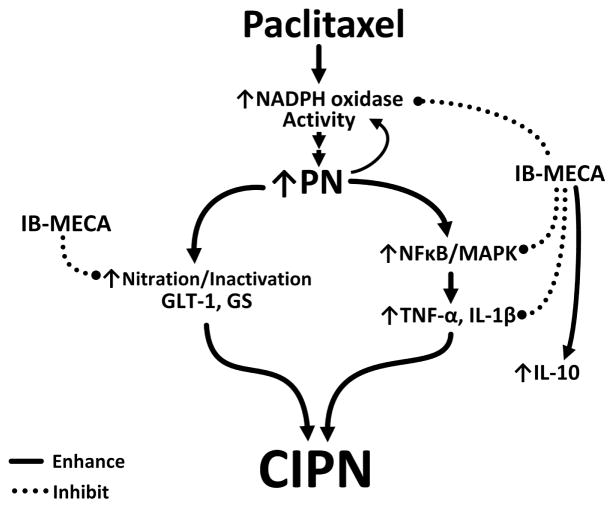
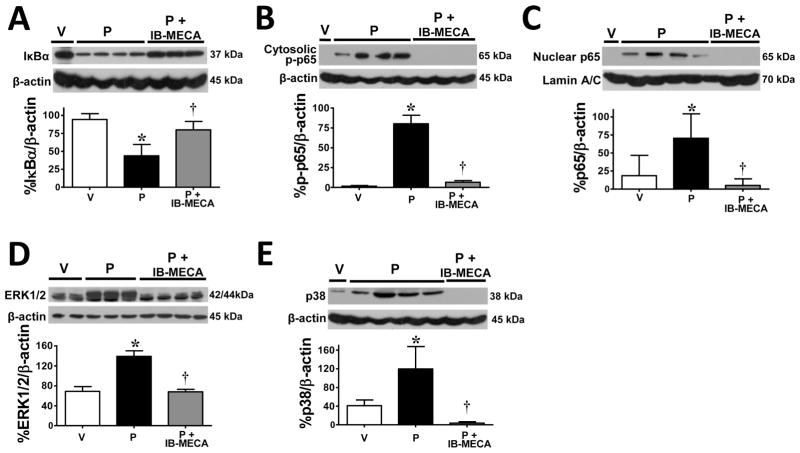
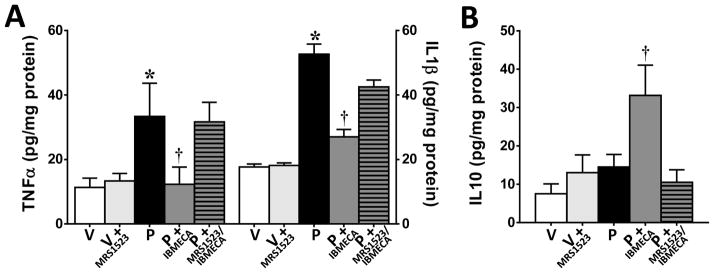
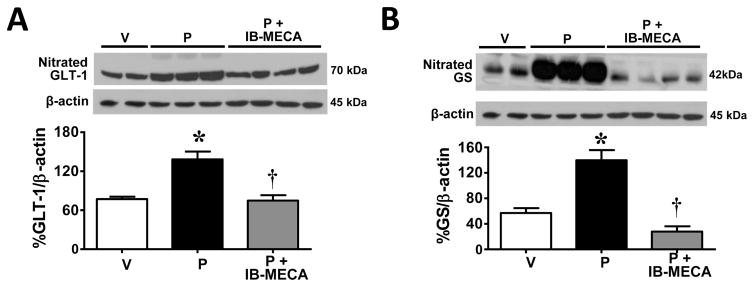
Chemotherapy-induced peripheral neuropathy accompanied by chronic neuropathic pain is the major dose-limiting toxicity of several anticancer agents including the taxane paclitaxel (Taxol). A critical mechanism underlying paclitaxel-induced neuropathic pain is the increased production of peroxynitrite in spinal cord generated in response to activation of the superoxide-generating enzyme, NADPH oxidase. Peroxynitrite in turn contributes to the development of neuropathic pain by modulating several redox-dependent events in spinal cord. We recently reported that activation of the Gi/Gq-coupled A3 adenosine receptor (A3AR) with selective A3AR agonists (ie, IB-MECA) blocked the development of chemotherapy induced-neuropathic pain evoked by distinct agents, including paclitaxel, without interfering with anticancer effects. The mechanism or mechanisms of action underlying these beneficial effects has yet to be explored. We now demonstrate that IB-MECA attenuates the development of paclitaxel-induced neuropathic pain by inhibiting the activation of spinal NADPH oxidase and two downstream redox-dependent systems. The first relies on inhibition of the redox-sensitive transcription factor (NFκB) and mitogen activated protein kinases (ERK and p38) resulting in decreased production of neuroexcitatory/proinflammatory cytokines (TNF-α, IL-1β) and increased formation of the neuroprotective/anti-inflammatory IL-10. The second involves inhibition of redox-mediated posttranslational tyrosine nitration and modification (inactivation) of glia-restricted proteins known to play key roles in regulating synaptic glutamate homeostasis: the glutamate transporter GLT-1 and glutamine synthetase. Our results unravel a mechanistic link into biomolecular signaling pathways employed by A3AR activation in neuropathic pain while providing the foundation to consider use of A3AR agonists as therapeutic agents in patients with chemotherapy-induced peripheral neuropathy.
化疗诱导的周围神经病变伴慢性神经性疼痛是包括紫杉烷类紫杉醇(泰素)在内的多种抗癌药物的主要剂量限制性毒性。紫杉醇诱导神经性疼痛的一个关键机制是,超氧化物生成酶NADPH氧化酶激活后,脊髓中过氧亚硝酸盐的生成增加。过氧亚硝酸盐进而通过调节脊髓中几个氧化还原依赖性事件,促进神经性疼痛的发展。我们最近报告称,用选择性A3腺苷受体(A3AR)激动剂(即IB-MECA)激活Gi/Gq偶联的A3AR,可阻断包括紫杉醇在内的不同药物诱发的化疗诱导神经性疼痛的发展,且不干扰抗癌效果。这些有益作用的作用机制尚待探索。我们现在证明,IB-MECA通过抑制脊髓NADPH氧化酶和两个下游氧化还原依赖性系统的激活,减轻紫杉醇诱导神经性疼痛的发展。第一个系统依赖于对氧化还原敏感的转录因子(NFκB)和丝裂原活化蛋白激酶(ERK和p38)的抑制,导致神经兴奋性/促炎细胞因子(TNF-α、IL-1β)的产生减少,以及神经保护性/抗炎性IL-10的生成增加。第二个系统涉及抑制氧化还原介导的翻译后酪氨酸硝化以及对已知在调节突触谷氨酸稳态中起关键作用的胶质细胞限制性蛋白的修饰(失活):谷氨酸转运体GLT-1和谷氨酰胺合成酶。我们的结果揭示了A3AR激活在神经性疼痛中所采用的生物分子信号通路的机制联系,同时为考虑将A3AR激动剂用作化疗诱导周围神经病变患者的治疗药物提供了基础。