BMC Bioinformatics. 2014;15 Suppl 9(Suppl 9):S4. doi: 10.1186/1471-2105-15-S9-S4. Epub 2014 Sep 10.
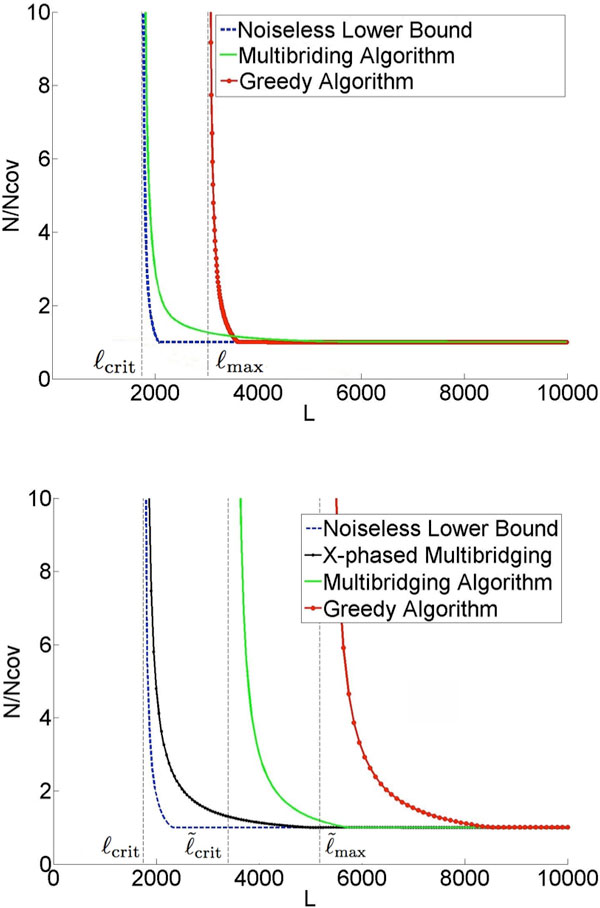
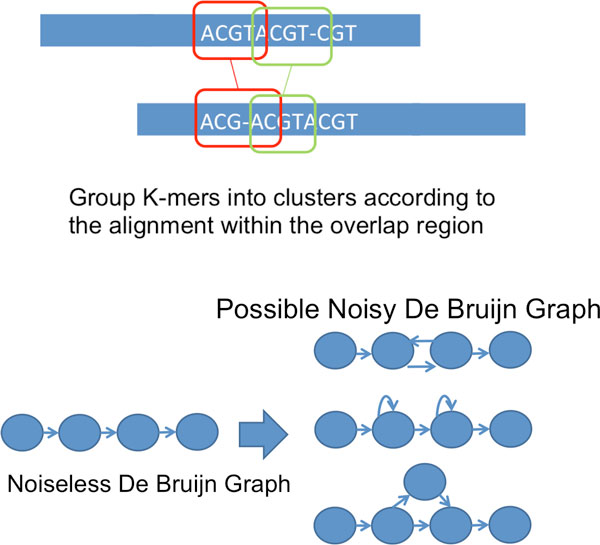
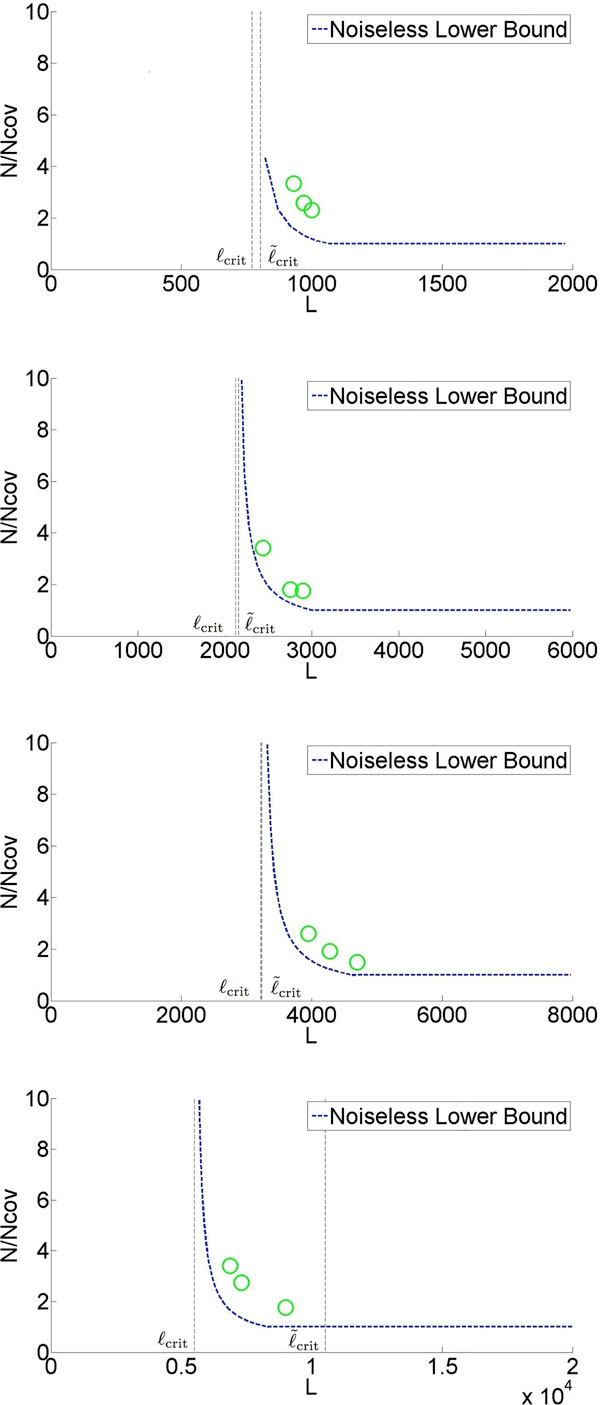
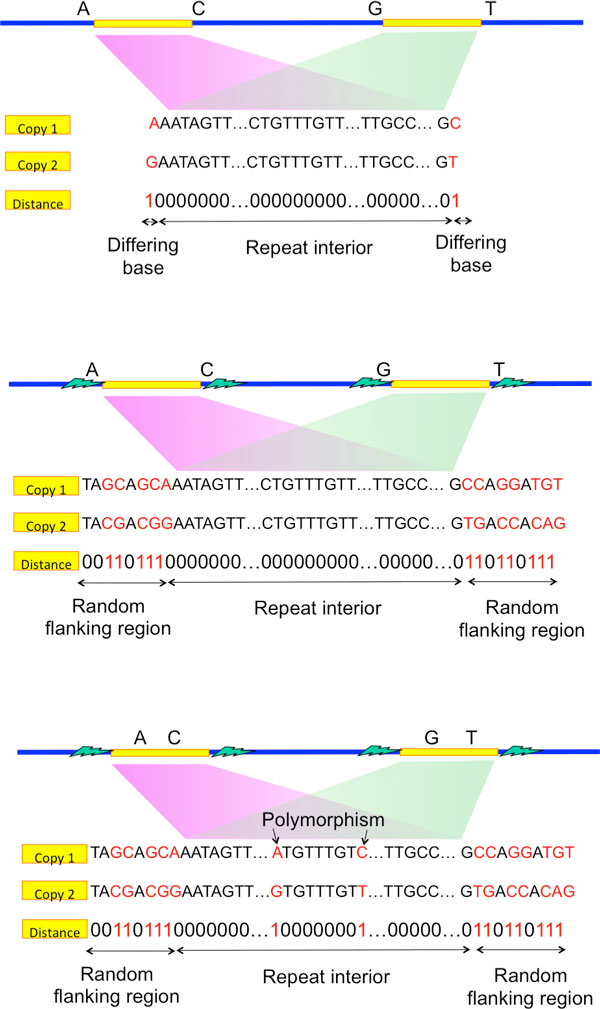
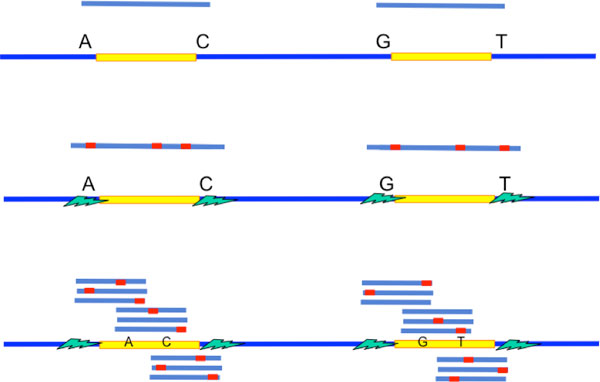
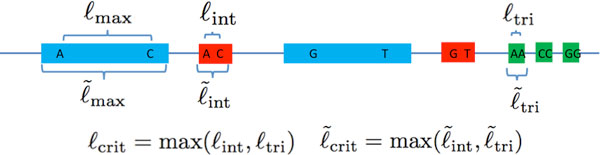
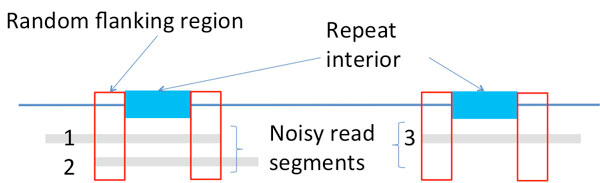
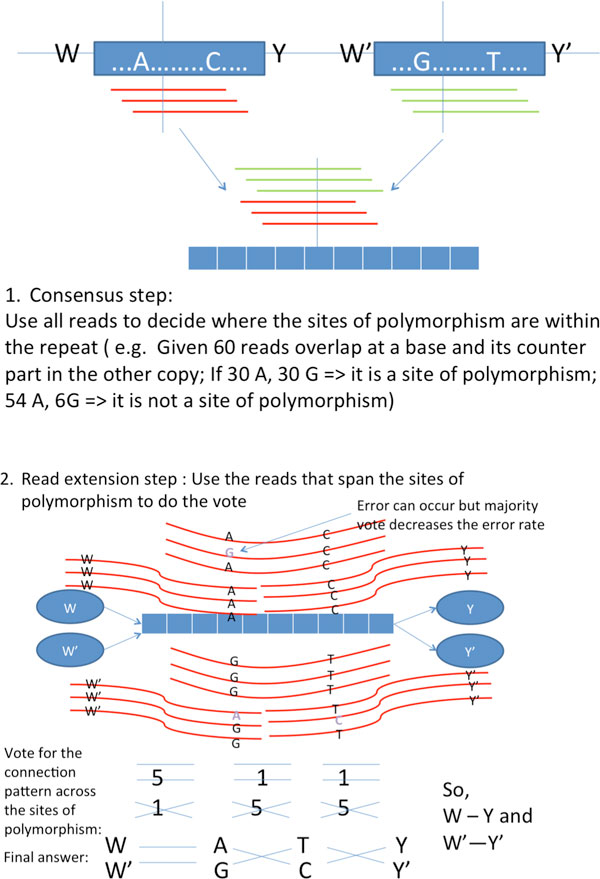
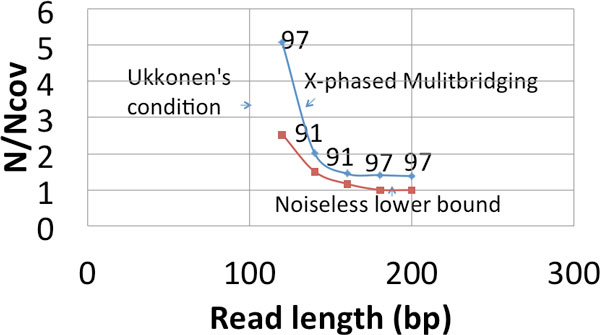
Recent work identified the fundamental limits on the information requirements in terms of read length and coverage depth required for successful de novo genome reconstruction from shotgun sequencing data, based on the idealistic assumption of no errors in the reads (noiseless reads). In this work, we show that even when there is noise in the reads, one can successfully reconstruct with information requirements close to the noiseless fundamental limit. A new assembly algorithm, X-phased Multibridging, is designed based on a probabilistic model of the genome. It is shown through analysis to perform well on the model, and through simulations to perform well on real genomes.
最近的工作确定了从鸟枪法测序数据成功从头重建基因组所需的读长和覆盖深度的信息要求的基本限制,这是基于读取中没有错误(无噪声读取)的理想化假设。在这项工作中,我们表明,即使读取中有噪声,也可以接近无噪声基本限制成功地进行重建。基于基因组的概率模型设计了一种新的组装算法 X 相多桥接。通过对模型的分析表明该算法性能良好,通过对真实基因组的模拟也表明该算法性能良好。