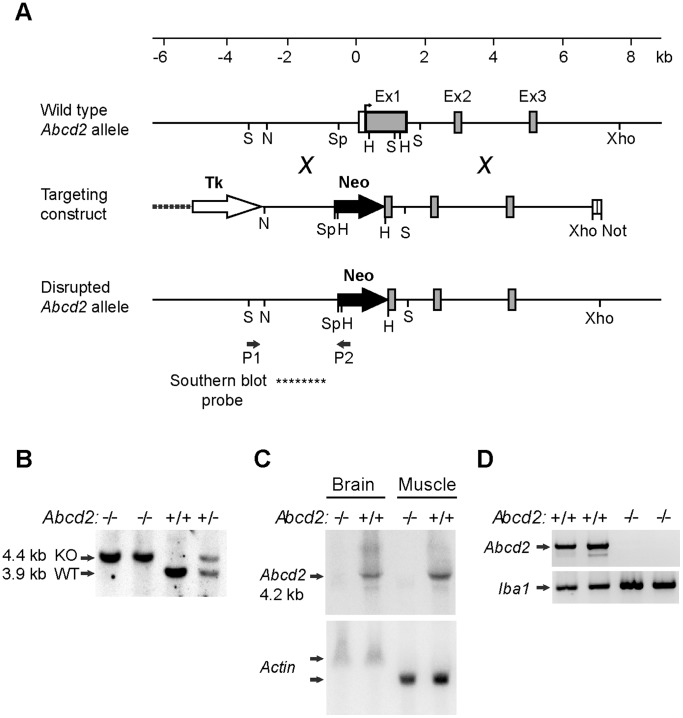
Muneer Zahid, Wiesinger Christoph, Voigtländer Till, Werner Hauke B, Berger Johannes, Forss-Petter Sonja
Center for Brain Research, Medical University of Vienna, Vienna, Austria.
Institute of Neurology, Medical University of Vienna, Vienna, Austria.
PLoS One. 2014 Sep 25;9(9):e108655. doi: 10.1371/journal.pone.0108655. eCollection 2014.
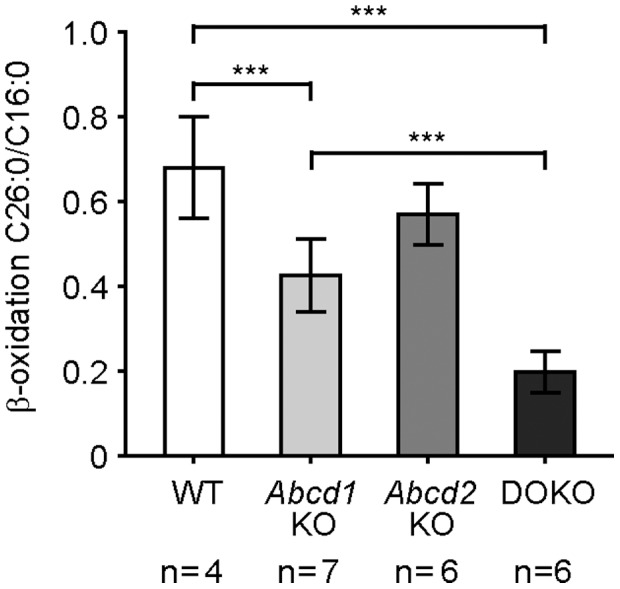
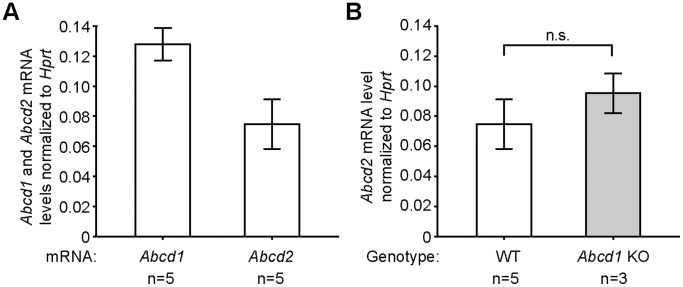
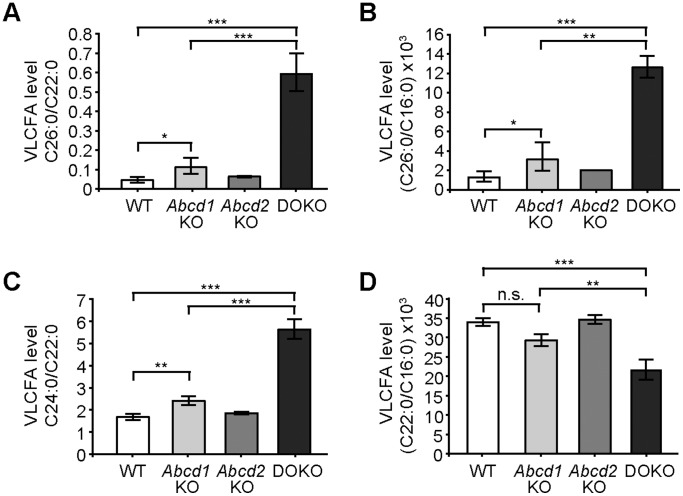
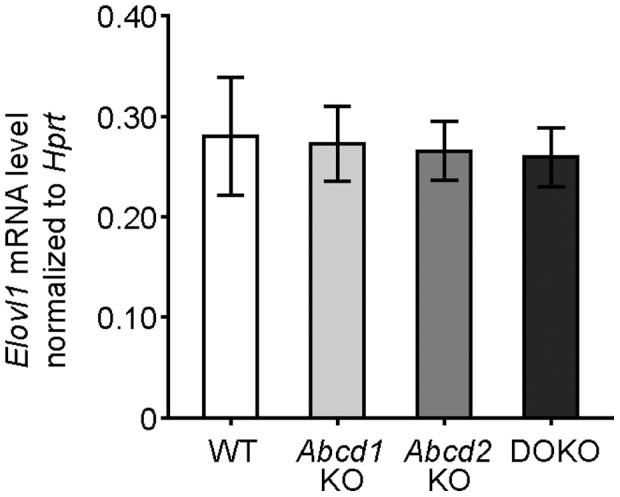
The inherited peroxisomal disorder X-linked adrenoleukodystrophy (X-ALD), associated with neurodegeneration and inflammatory cerebral demyelination, is caused by mutations in the ABCD1 gene encoding the peroxisomal ATP-binding cassette (ABC) transporter ABCD1 (ALDP). ABCD1 transports CoA-esters of very long-chain fatty acids (VLCFA) into peroxisomes for degradation by β-oxidation; thus, ABCD1 deficiency results in VLCFA accumulation. The closest homologue, ABCD2 (ALDRP), when overexpressed, compensates for ABCD1 deficiency in X-ALD fibroblasts and in Abcd1-deficient mice. Microglia/macrophages have emerged as important players in the progression of neuroinflammation. Human monocytes, lacking significant expression of ABCD2, display severely impaired VLCFA metabolism in X-ALD. Here, we used thioglycollate-elicited primary mouse peritoneal macrophages (MPMΦ) from Abcd1 and Abcd2 single- and double-deficient mice to establish how these mutations affect VLCFA metabolism. By quantitative RT-PCR, Abcd2 mRNA was about half as abundant as Abcd1 mRNA in wild-type and similarly abundant in Abcd1-deficient MPMΦ. VLCFA (C26∶0) accumulated about twofold in Abcd1-deficient MPMΦ compared with wild-type controls, as measured by gas chromatography-mass spectrometry. In Abcd2-deficient macrophages VLCFA levels were normal. However, upon Abcd1/Abcd2 double-deficiency, VLCFA accumulation was markedly increased (sixfold) compared with Abcd1-deficient MPMΦ. Elovl1 mRNA, encoding the rate-limiting enzyme for elongation of VLCFA, was equally abundant across all genotypes. Peroxisomal β-oxidation of C26∶0 amounted to 62% of wild-type activity in Abcd1-deficient MPMΦ and was significantly more impaired (29% residual activity) upon Abcd1/Abcd2 double-deficiency. Single Abcd2 deficiency did not significantly compromise β-oxidation of C26∶0. Thus, the striking accumulation of VLCFA in double-deficient MPMΦ compared with single Abcd1 deficiency was due to the loss of ABCD2-mediated, compensatory transport of VLCFA into peroxisomes. We propose that moderate endogenous expression of Abcd2 in Abcd1-deficient murine macrophages prevents the severe metabolic phenotype observed in human X-ALD monocytes, which lack appreciable expression of ABCD2. This supports upregulation of ABCD2 as a therapeutic concept in X-ALD.
遗传性过氧化物酶体疾病X连锁肾上腺脑白质营养不良(X-ALD)与神经退行性变和炎症性脑脱髓鞘相关,由编码过氧化物酶体ATP结合盒(ABC)转运蛋白ABCD1(ALDP)的ABCD1基因突变引起。ABCD1将极长链脂肪酸(VLCFA)的辅酶A酯转运到过氧化物酶体中,通过β氧化进行降解;因此,ABCD1缺乏会导致VLCFA积累。最接近的同源物ABCD2(ALDRP)在过表达时,可补偿X-ALD成纤维细胞和Abcd1缺陷小鼠中的ABCD1缺乏。小胶质细胞/巨噬细胞已成为神经炎症进展中的重要参与者。缺乏ABCD2显著表达的人类单核细胞在X-ALD中表现出严重受损的VLCFA代谢。在这里,我们使用来自Abcd1和Abcd2单缺陷和双缺陷小鼠的巯基乙酸诱导的原代小鼠腹腔巨噬细胞(MPMΦ)来确定这些突变如何影响VLCFA代谢。通过定量RT-PCR,在野生型中Abcd2 mRNA的丰度约为Abcd1 mRNA的一半,在Abcd1缺陷的MPMΦ中丰度相似。通过气相色谱-质谱法测量,与野生型对照相比,Abcd1缺陷的MPMΦ中VLCFA(C26∶0)积累约两倍。在Abcd2缺陷的巨噬细胞中,VLCFA水平正常。然而,在Abcd1/Abcd2双缺陷时,与Abcd1缺陷的MPMΦ相比,VLCFA积累显著增加(六倍)。编码VLCFA延长限速酶的Elovl1 mRNA在所有基因型中丰度相同。在Abcd1缺陷的MPMΦ中,C26∶0的过氧化物酶体β氧化相当于野生型活性的62%,在Abcd1/Abcd2双缺陷时受损更严重(残余活性29%)。单一Abcd2缺陷不会显著损害C26∶0的β氧化。因此,与单一Abcd1缺陷相比,双缺陷MPMΦ中VLCFA的显著积累是由于失去了ABCD2介导的VLCFA向过氧化物酶体的代偿性转运。我们提出,在Abcd1缺陷的小鼠巨噬细胞中Abcd2的适度内源性表达可防止在缺乏ABCD2明显表达的人类X-ALD单核细胞中观察到的严重代谢表型。这支持将上调ABCD2作为X-ALD的一种治疗理念。