Bouwman Aniek C, Veerkamp Roel F
Animal Breeding and Genomics Centre, Wageningen UR Livestock Research, P.O. Box 338, 6700, AH, Wageningen, Netherlands.
BMC Genet. 2014 Oct 3;15:105. doi: 10.1186/s12863-014-0105-8.
The aim of this study was to determine the consequences of splitting sequencing effort over multiple breeds for imputation accuracy from a high-density SNP chip towards whole-genome sequence. Such information would assist for instance numerical smaller cattle breeds, but also pig and chicken breeders, who have to choose wisely how to spend their sequencing efforts over all the breeds or lines they evaluate. Sequence data from cattle breeds was used, because there are currently relatively many individuals from several breeds sequenced within the 1,000 Bull Genomes project. The advantage of whole-genome sequence data is that it carries the causal mutations, but the question is whether it is possible to impute the causal variants accurately. This study therefore focussed on imputation accuracy of variants with low minor allele frequency and breed specific variants.
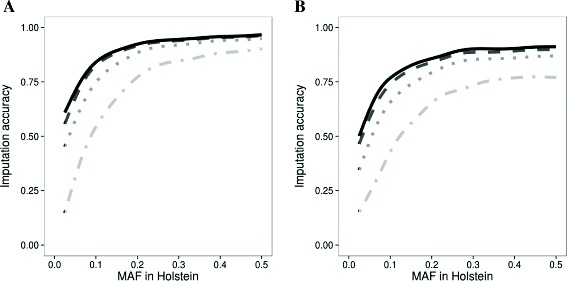
Imputation accuracy was assessed for chromosome 1 and 29 as the correlation between observed and imputed genotypes. For chromosome 1, the average imputation accuracy was 0.70 with a reference population of 20 Holstein, and increased to 0.83 when the reference population was increased by including 3 other dairy breeds with 20 animals each. When the same amount of animals from the Holstein breed were added the accuracy improved to 0.88, while adding the 3 other breeds to the reference population of 80 Holstein improved the average imputation accuracy marginally to 0.89. For chromosome 29, the average imputation accuracy was lower. Some variants benefitted from the inclusion of other breeds in the reference population, initially determined by the MAF of the variant in each breed, but even Holstein specific variants did gain imputation accuracy from the multi-breed reference population.
This study shows that splitting sequencing effort over multiple breeds and combining the reference populations is a good strategy for imputation from high-density SNP panels towards whole-genome sequence when reference populations are small and sequencing effort is limiting. When sequencing effort is limiting and interest lays in multiple breeds or lines this provides imputation of each breed.
本研究的目的是确定将测序工作分散到多个品种上对从高密度SNP芯片向全基因组序列进行推算准确性的影响。此类信息将有助于例如数量较少的牛品种,以及猪和鸡的育种者,他们必须明智地选择如何在他们评估的所有品种或品系上分配测序工作。使用了牛品种的序列数据,因为目前在1000公牛基因组计划中有来自几个品种的相对较多的个体进行了测序。全基因组序列数据的优势在于它携带了因果突变,但问题是是否有可能准确推算出因果变异。因此,本研究重点关注低小等位基因频率变异和品种特异性变异的推算准确性。
以观察到的基因型与推算出的基因型之间的相关性为指标,评估了第1号和第29号染色体的推算准确性。对于第1号染色体,当参考群体为20头荷斯坦奶牛时,平均推算准确性为0.70,当参考群体增加另外3个各有20头动物的奶牛品种时,准确性提高到0.83。当加入相同数量的荷斯坦奶牛品种个体时,准确性提高到0.88,而在80头荷斯坦奶牛的参考群体中加入另外3个品种,平均推算准确性略有提高,达到0.89。对于第29号染色体,平均推算准确性较低。一些变异从参考群体中纳入其他品种中受益,最初由每个品种中变异的小等位基因频率决定,但即使是荷斯坦奶牛特有的变异也从多品种参考群体中提高了推算准确性。
本研究表明,当参考群体较小且测序工作有限时,将测序工作分散到多个品种并合并参考群体是从高密度SNP面板向全基因组序列进行推算的一个好策略。当测序工作有限且关注多个品种或品系时,这可以对每个品种进行推算。