Fiocchetti M, Nuzzo M T, Totta P, Acconcia F, Ascenzi P, Marino M
Department of Science, University Roma Tre, Viale Guglielmo Marconi 446, I-00146 Roma, Italy.
Interdepartmental Laboratory of Electron Microscopy, University Roma Tre, Via della Vasca Navale 79, I-00146 Roma, Italy.
Cell Death Dis. 2014 Oct 9;5(10):e1449. doi: 10.1038/cddis.2014.418.
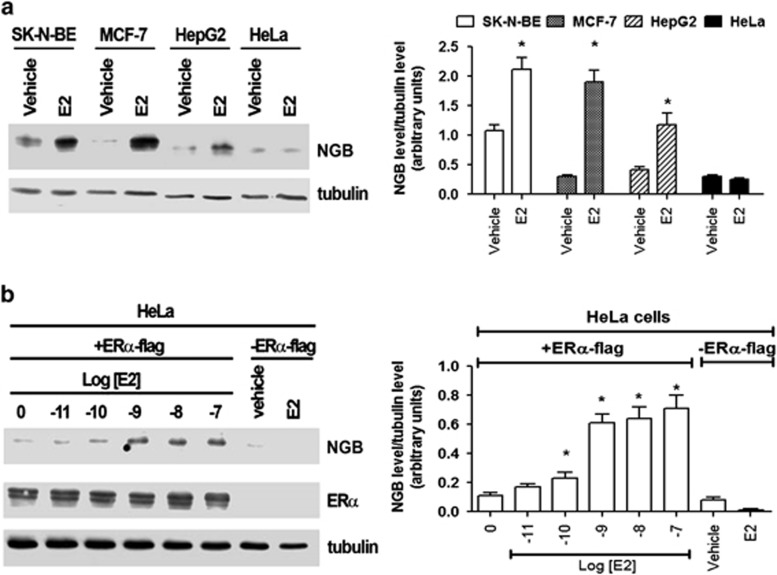
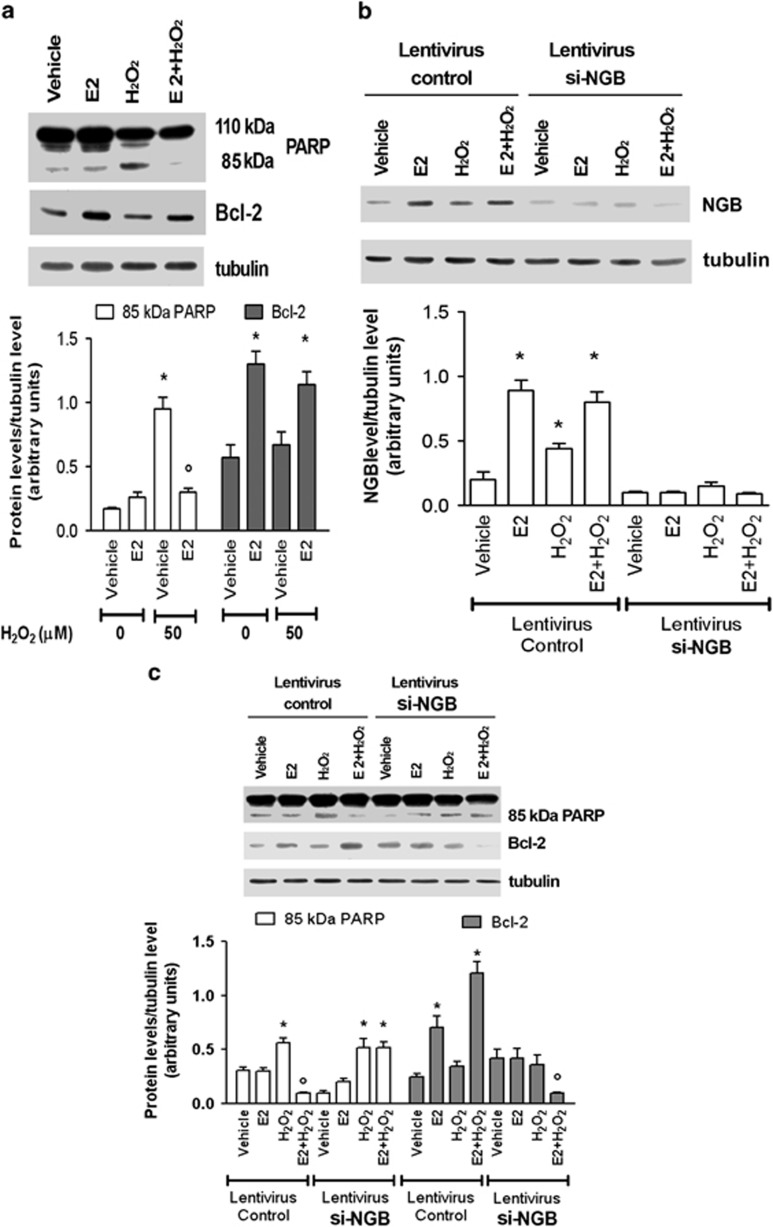
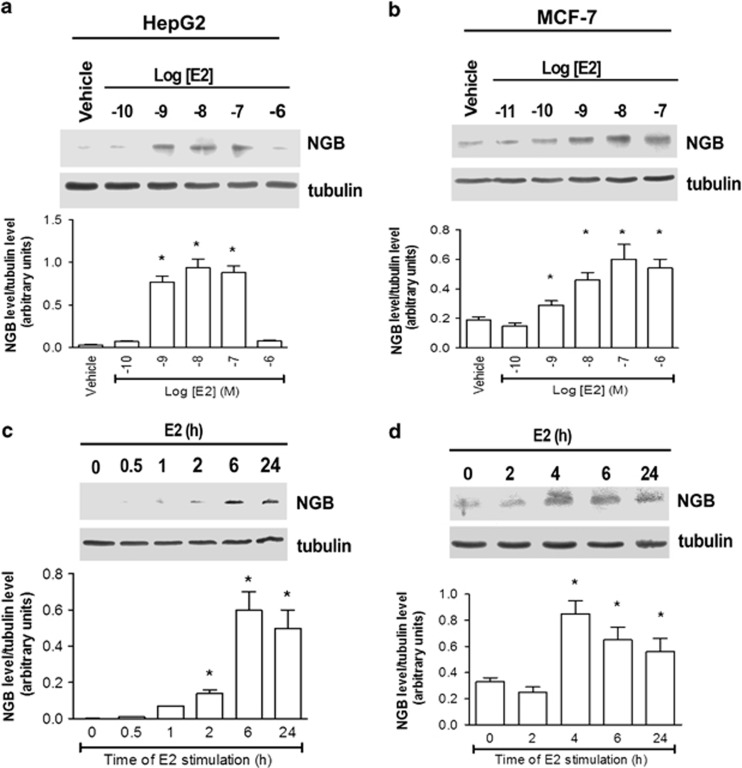
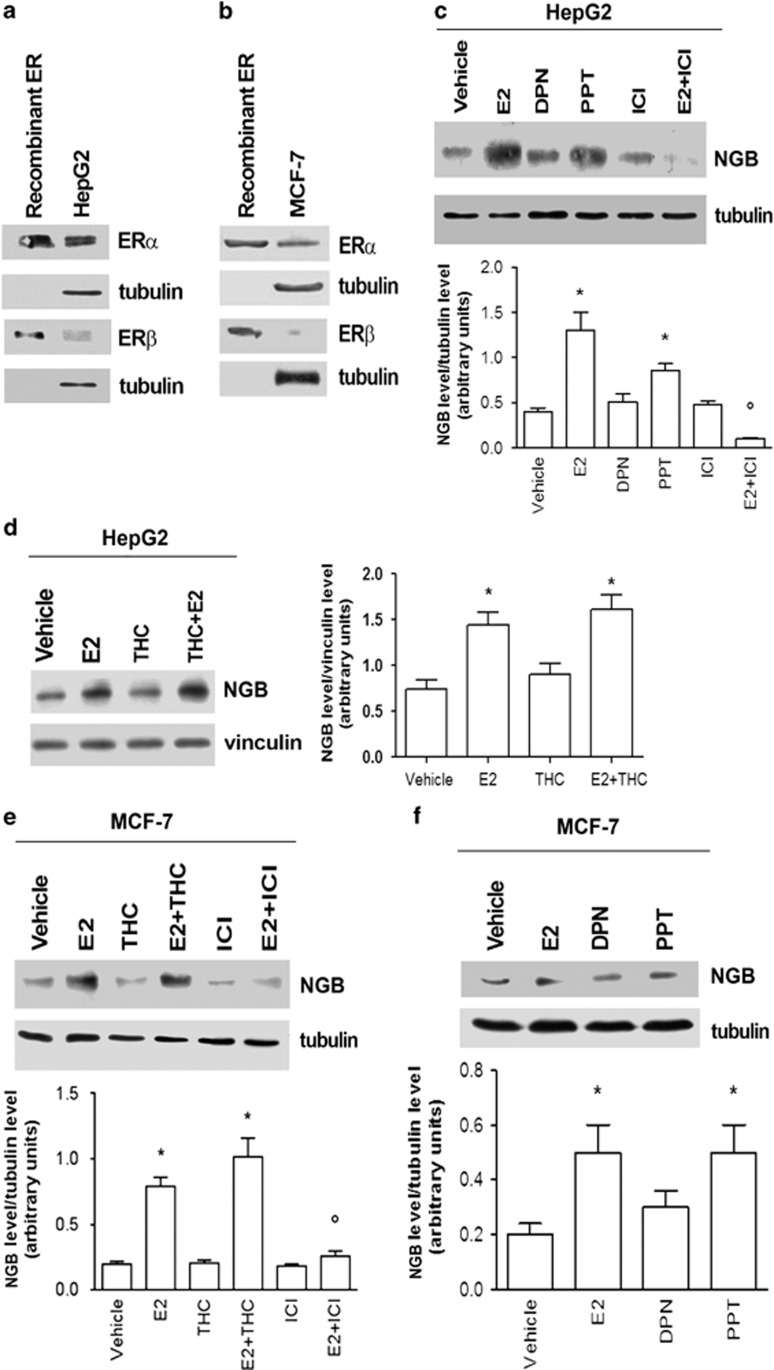
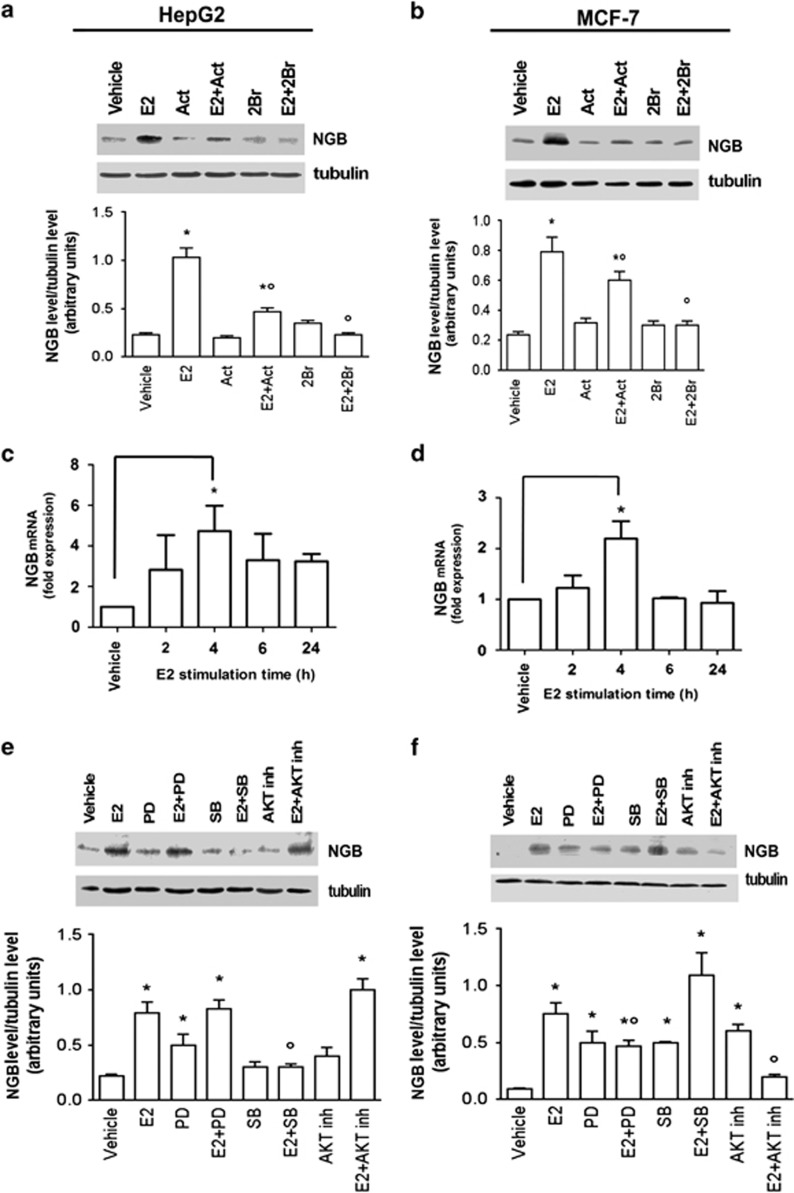
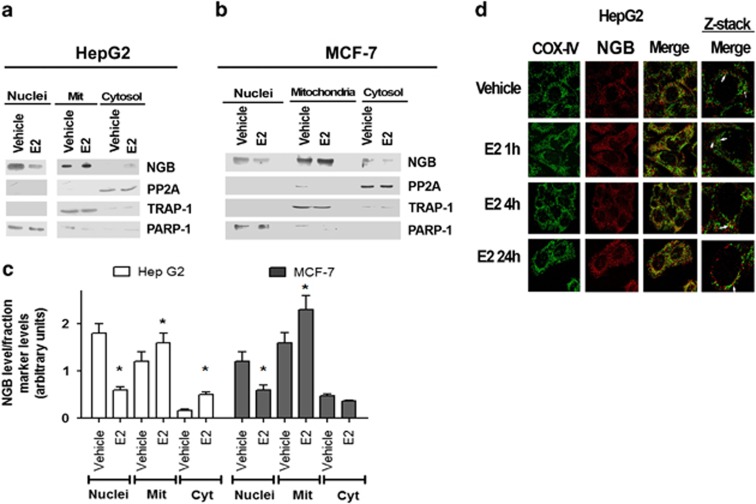
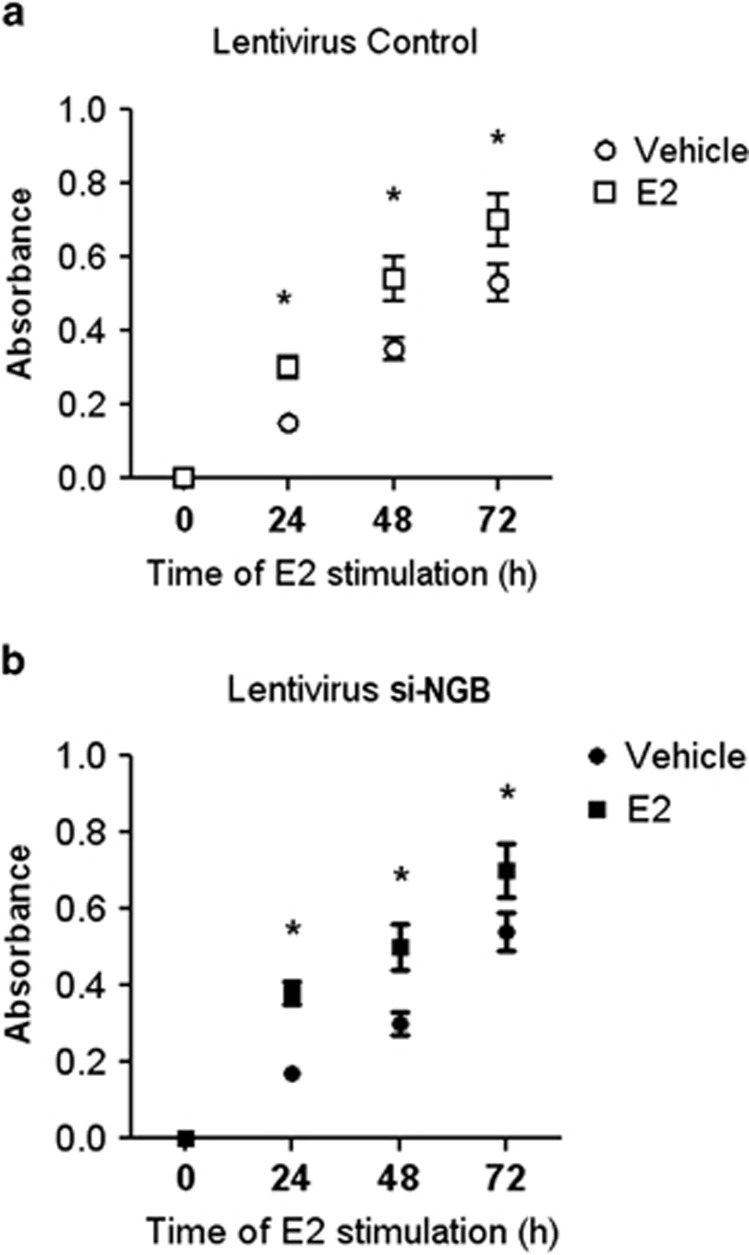
Recently, we reported that human neuroglobin (NGB) is a new player in the signal transduction pathways that lead to 17β-estradiol (E2)-induced neuron survival. Indeed, E2 induces in neuron mitochondria the enhancement of NGB level, which in turn impairs the activation of a pro-apoptotic cascade. Nowadays, the existence of a similar pathway activated by E2 in non-neuronal cells is completely unknown. Here, the role of E2-induced NGB upregulation in tumor cells is reported. E2 induced the upregulation of NGB in a dose- and time-dependent manner in MCF-7, HepG2, SK-N-BE, and HeLa cells transfected with estrogen receptor α (ERα), whereas E2 was unable to modulate the NGB expression in the ERα-devoid HeLa cells. Both transcriptional and extranuclear ERα signals were required for the E2-dependent upregulation of NGB in MCF-7 and HepG2 cell lines. E2 stimulation modified NGB intracellular localization, inducing a significant reduction of NGB in the nucleus with a parallel increase of NGB in the mitochondria in both HepG2 and MCF-7 cells. Remarkably, E2 pretreatment did not counteract the H2O2-induced caspase-3 and poly (ADP-ribose) polymerase 1 (PARP-1) cleavage, as well as Bcl-2 overexpression in MCF-7 and HepG2 cells in which NGB was stably silenced by using shRNA lentiviral particles, highlighting the pivotal role of NGB in E2-induced antiapoptotic pathways in cancer cells. Present results indicate that the E2-induced NGB upregulation in cancer cells could represent a defense mechanism of E2-related cancers rendering them insensitive to oxidative stress. As a whole, these data open new avenues to develop therapeutic strategies against E2-related cancers.
最近,我们报道了人类神经球蛋白(NGB)是导致17β-雌二醇(E2)诱导神经元存活的信号转导途径中的一个新成员。事实上,E2可诱导神经元线粒体中NGB水平升高,进而损害促凋亡级联反应的激活。目前,E2在非神经元细胞中激活的类似途径的存在情况完全未知。在此,我们报道了E2诱导的NGB上调在肿瘤细胞中的作用。E2以剂量和时间依赖性方式诱导转染了雌激素受体α(ERα)的MCF-7、HepG2、SK-N-BE和HeLa细胞中NGB上调,而E2无法调节缺乏ERα的HeLa细胞中NGB的表达。E2依赖性上调MCF-7和HepG2细胞系中的NGB需要转录和核外ERα信号。E2刺激改变了NGB的细胞内定位,导致HepG2和MCF-7细胞中细胞核内NGB显著减少,同时线粒体中NGB平行增加。值得注意的是,在使用慢病毒颗粒稳定沉默NGB的MCF-7和HepG2细胞中,E2预处理并未抵消H2O2诱导的半胱天冬酶-3和聚(ADP-核糖)聚合酶1(PARP-1)的切割以及Bcl-2的过表达,这突出了NGB在癌细胞中E2诱导的抗凋亡途径中的关键作用。目前的结果表明,E2诱导癌细胞中NGB上调可能代表E2相关癌症的一种防御机制,使其对氧化应激不敏感。总体而言,这些数据为开发针对E2相关癌症的治疗策略开辟了新途径。