Andrews Logan D, Zalatan Jesse G, Herschlag Daniel
Department of Chemical and Systems Biology, ‡Department of Chemistry, and §Department of Biochemistry, Stanford University , Stanford, California 94305-5307, United States.
Biochemistry. 2014 Nov 4;53(43):6811-9. doi: 10.1021/bi500765p. Epub 2014 Oct 23.
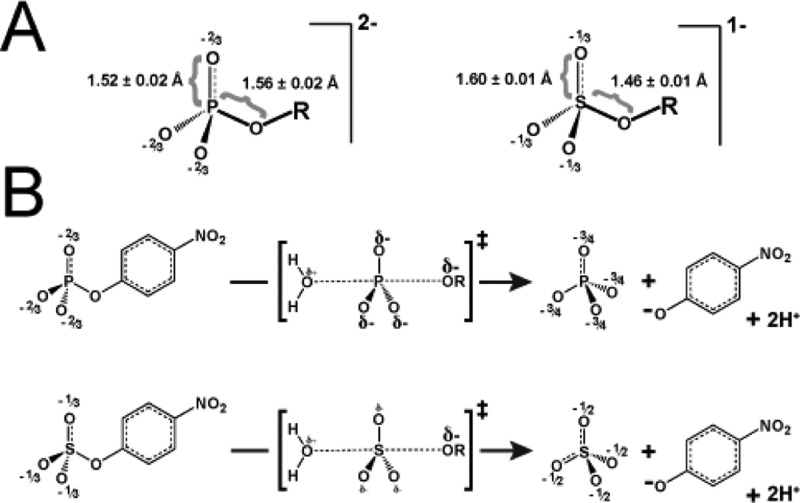
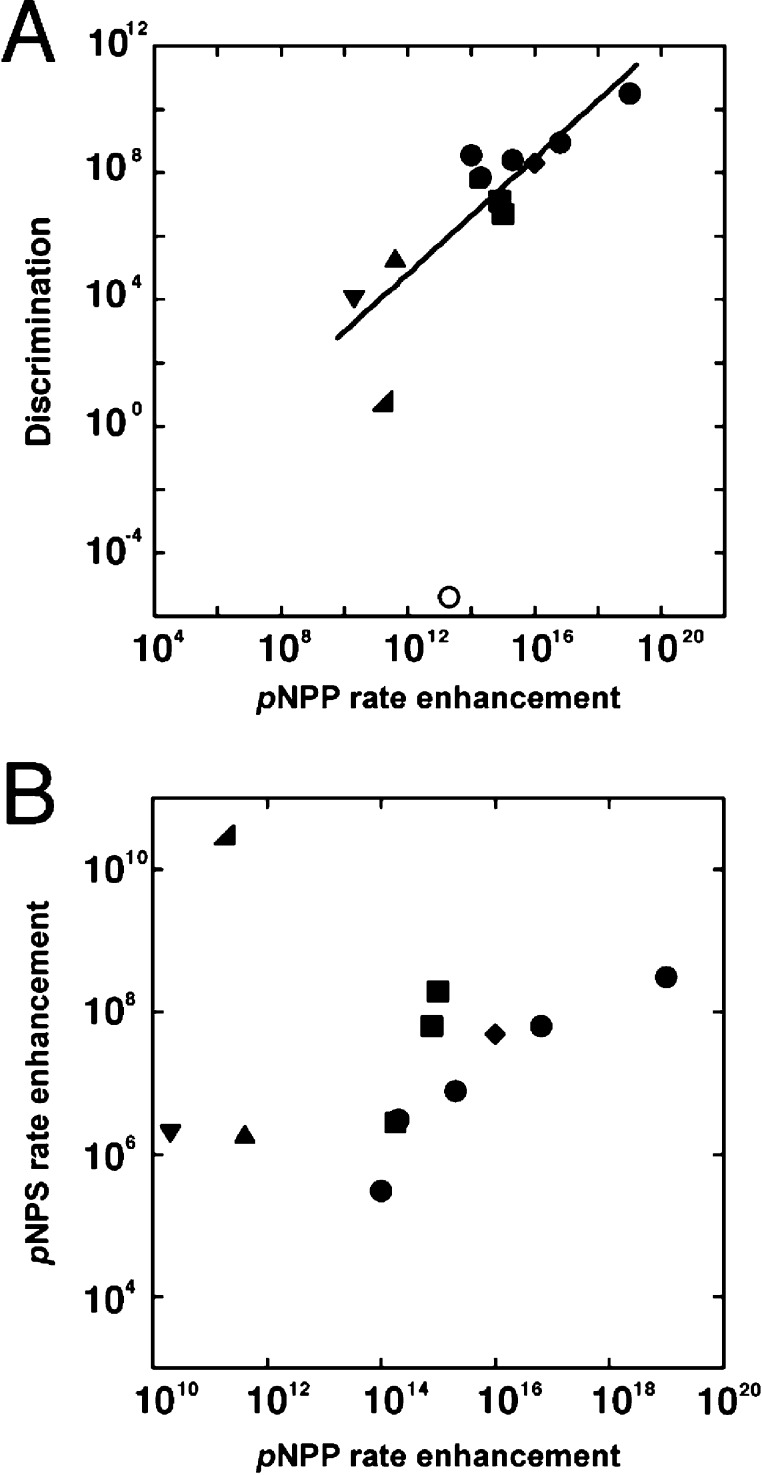
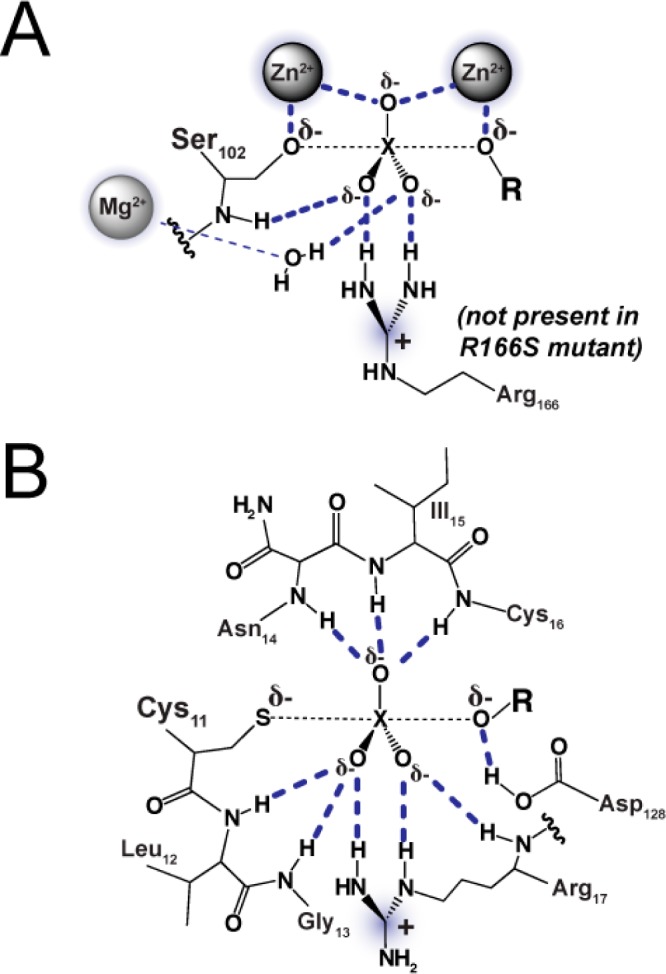
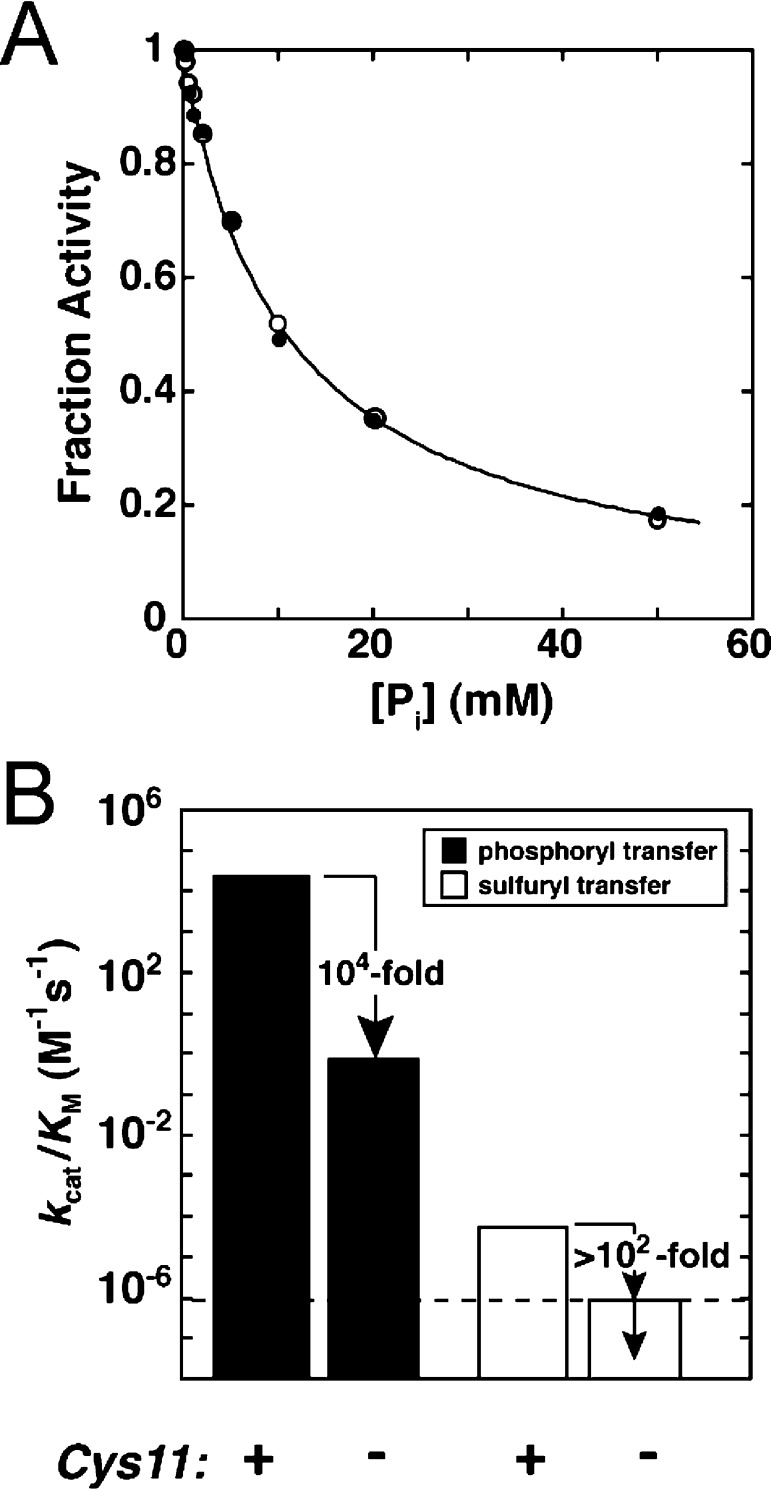
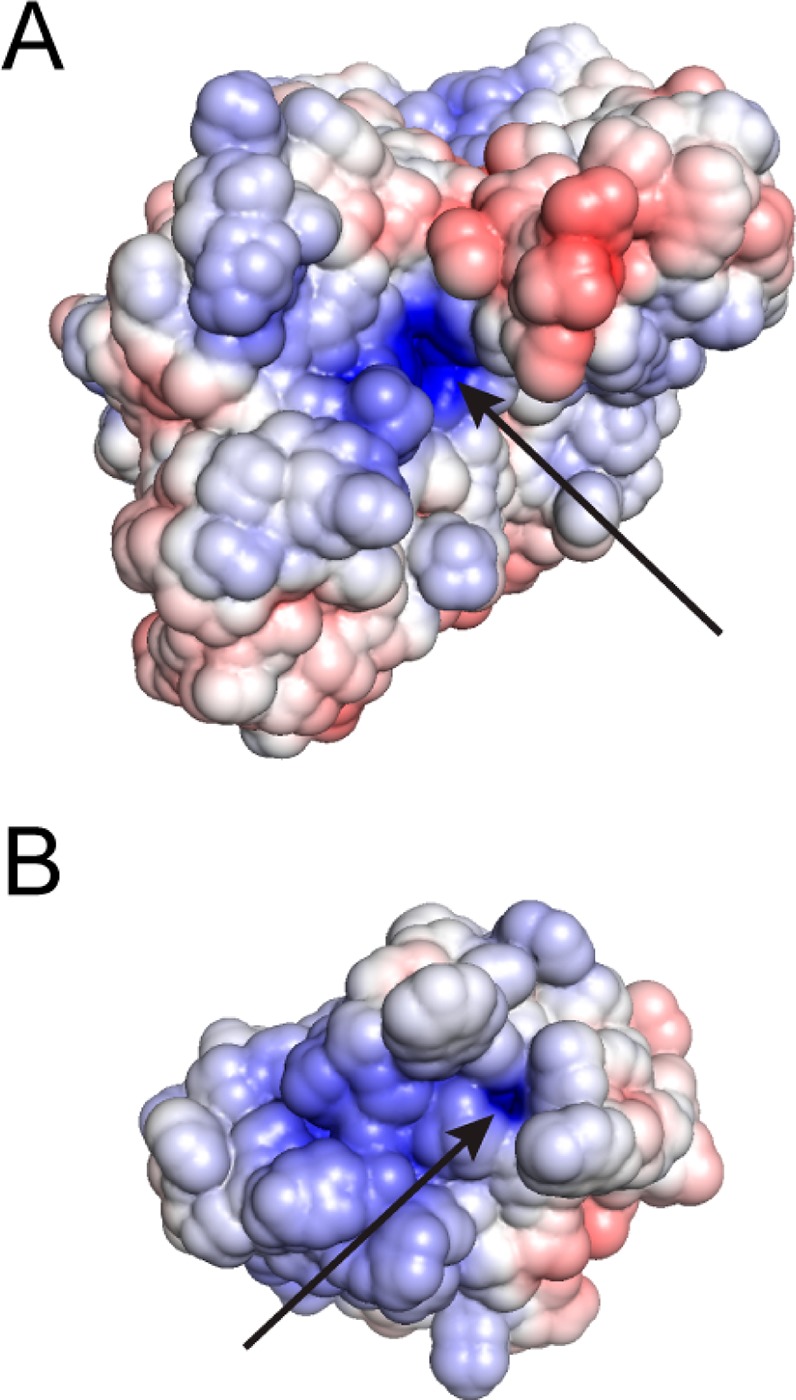
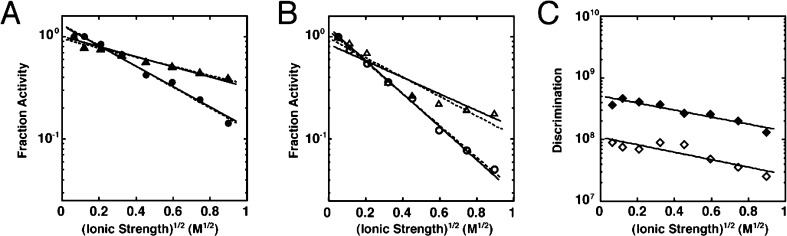
Catalytic promiscuity, the ability of enzymes to catalyze multiple reactions, provides an opportunity to gain a deeper understanding of the origins of catalysis and substrate specificity. Alkaline phosphatase (AP) catalyzes both phosphate and sulfate monoester hydrolysis reactions with a ∼10(10)-fold preference for phosphate monoester hydrolysis, despite the similarity between these reactions. The preponderance of formal positive charge in the AP active site, particularly from three divalent metal ions, was proposed to be responsible for this preference by providing stronger electrostatic interactions with the more negatively charged phosphoryl group versus the sulfuryl group. To test whether positively charged metal ions are required to achieve a high preference for the phosphate monoester hydrolysis reaction, the catalytic preference of three protein tyrosine phosphatases (PTPs), which do not contain metal ions, were measured. Their preferences ranged from 5 × 10(6) to 7 × 10(7), lower than that for AP but still substantial, indicating that metal ions and a high preponderance of formal positive charge within the active site are not required to achieve a strong catalytic preference for phosphate monoester over sulfate monoester hydrolysis. The observed ionic strength dependences of kcat/KM values for phosphate and sulfate monoester hydrolysis are steeper for the more highly charged phosphate ester with both AP and the PTP Stp1, following the dependence expected based on the charge difference of these two substrates. However, the dependences for AP were not greater than those of Stp1 and were rather shallow for both enzymes. These results suggest that overall electrostatics from formal positive charge within the active site is not the major driving force in distinguishing between these reactions and that substantial discrimination can be attained without metal ions. Thus, local properties of the active site, presumably including multiple positioned dipolar hydrogen bond donors within the active site, dominate in defining this reaction specificity.
催化多效性,即酶催化多种反应的能力,为更深入理解催化作用和底物特异性的起源提供了契机。碱性磷酸酶(AP)能催化磷酸单酯和硫酸单酯的水解反应,尽管这两种反应相似,但AP对磷酸单酯水解的偏好性约为10¹⁰倍。AP活性位点中大量的形式正电荷,特别是来自三个二价金属离子,被认为是造成这种偏好性的原因,因为与带更多负电荷的磷酰基相比,它们与硫酰基的静电相互作用更强。为了测试带正电荷的金属离子是否是实现对磷酸单酯水解反应高度偏好所必需的,我们测量了三种不含金属离子的蛋白质酪氨酸磷酸酶(PTP)的催化偏好性。它们的偏好性范围在5×10⁶到7×10⁷之间,低于AP,但仍然相当可观,这表明活性位点内的金属离子和大量形式正电荷并非实现对磷酸单酯水解比对硫酸单酯水解有强烈催化偏好所必需的。对于AP和PTP Stp1,随着离子强度的增加,磷酸单酯和硫酸单酯水解的kcat/KM值对离子强度的依赖性,对于电荷更高的磷酸酯更为陡峭,这符合基于这两种底物电荷差异所预期的依赖性。然而,AP的依赖性并不比Stp1的大,而且两种酶的依赖性都相当浅。这些结果表明,活性位点内形式正电荷产生的整体静电作用并非区分这些反应的主要驱动力,并且在没有金属离子的情况下也能实现显著的区分。因此,活性位点的局部性质,可能包括活性位点内多个定位的偶极氢键供体,在定义这种反应特异性中起主导作用。