Chaki Mounira, Kovacs Izabella, Spannagl Manuel, Lindermayr Christian
Institute of Biochemical Plant Pathology, Helmholtz Zentrum München-German Research Center for Environmental Health, Neuherberg, Germany.
Institute of Bioinformatics and Systems Biology, Helmholtz Zentrum München-German Research Center for Environmental Health, Neuherberg, Germany.
PLoS One. 2014 Oct 21;9(10):e110232. doi: 10.1371/journal.pone.0110232. eCollection 2014.
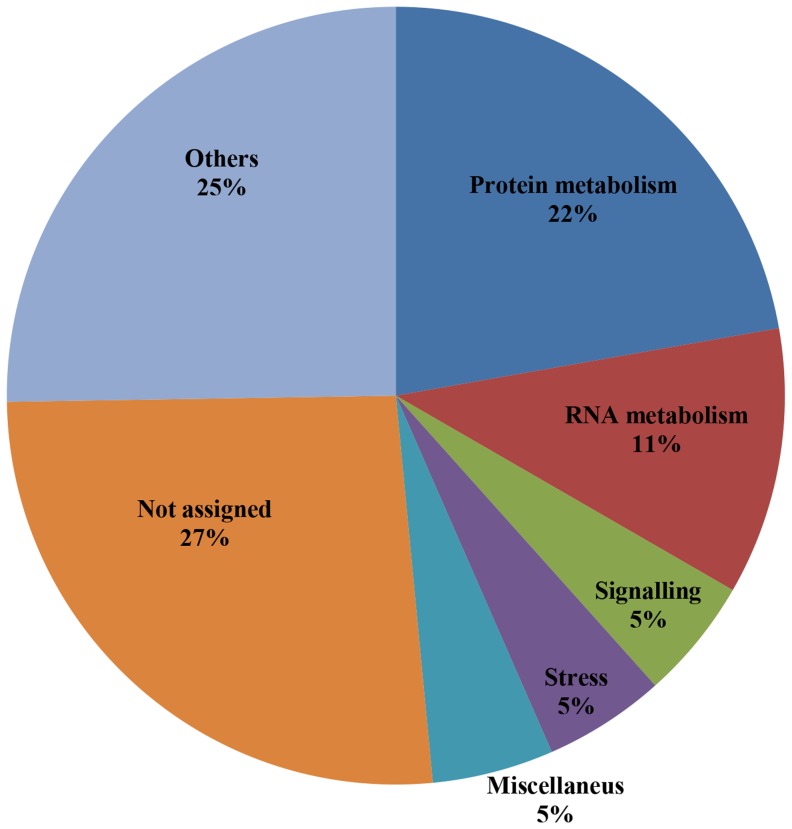
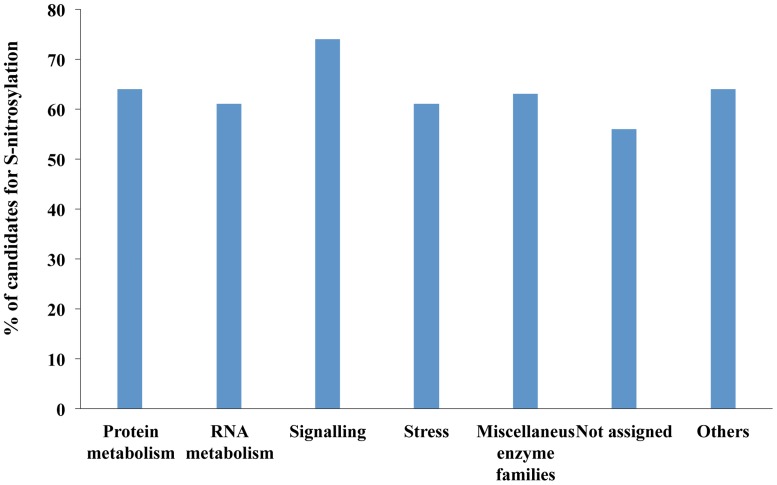
Nitric oxide (NO) is an important signaling molecule that regulates many physiological processes in plants. One of the most important regulatory mechanisms of NO is S-nitrosylation-the covalent attachment of NO to cysteine residues. Although the involvement of cysteine S-nitrosylation in the regulation of protein functions is well established, its substrate specificity remains unknown. Identification of candidates for S-nitrosylation and their target cysteine residues is fundamental for studying the molecular mechanisms and regulatory roles of S-nitrosylation in plants. Several experimental methods that are based on the biotin switch have been developed to identify target proteins for S-nitrosylation. However, these methods have their limits. Thus, computational methods are attracting considerable attention for the identification of modification sites in proteins. Using GPS-SNO version 1.0, a recently developed S-nitrosylation site-prediction program, a set of 16,610 candidate proteins for S-nitrosylation containing 31,900 S-nitrosylation sites was isolated from the entire Arabidopsis proteome using the medium threshold. In the compartments "chloroplast," "CUL4-RING ubiquitin ligase complex," and "membrane" more than 70% of the proteins were identified as candidates for S-nitrosylation. The high number of identified candidates in the proteome reflects the importance of redox signaling in these compartments. An analysis of the functional distribution of the predicted candidates showed that proteins involved in signaling processes exhibited the highest prediction rate. In a set of 46 proteins, where 53 putative S-nitrosylation sites were already experimentally determined, the GPS-SNO program predicted 60 S-nitrosylation sites, but only 11 overlap with the results of the experimental approach. In general, a computer-assisted method for the prediction of targets for S-nitrosylation is a very good tool; however, further development, such as including the three dimensional structure of proteins in such analyses, would improve the identification of S-nitrosylation sites.
一氧化氮(NO)是一种重要的信号分子,可调节植物中的许多生理过程。NO最重要的调节机制之一是S-亚硝基化——NO与半胱氨酸残基的共价连接。尽管半胱氨酸S-亚硝基化参与蛋白质功能调节已得到充分证实,但其底物特异性仍然未知。鉴定S-亚硝基化的候选物及其靶半胱氨酸残基是研究植物中S-亚硝基化的分子机制和调节作用的基础。已经开发了几种基于生物素开关的实验方法来鉴定S-亚硝基化的靶蛋白。然而,这些方法有其局限性。因此,计算方法在蛋白质修饰位点的鉴定方面正引起相当大的关注。使用最近开发的S-亚硝基化位点预测程序GPS-SNO 1.0版本,通过中等阈值从整个拟南芥蛋白质组中分离出一组包含31900个S-亚硝基化位点的16610个S-亚硝基化候选蛋白。在“叶绿体”、“CUL4-RING泛素连接酶复合物”和“膜”区室中,超过70%的蛋白质被鉴定为S-亚硝基化候选物。蛋白质组中大量已鉴定的候选物反映了氧化还原信号在这些区室中的重要性。对预测候选物的功能分布分析表明,参与信号传导过程的蛋白质表现出最高的预测率。在一组46种蛋白质中,其中53个推定的S-亚硝基化位点已经通过实验确定,GPS-SNO程序预测了60个S-亚硝基化位点,但只有11个与实验方法的结果重叠。一般来说,一种用于预测S-亚硝基化靶标的计算机辅助方法是一个非常好的工具;然而,进一步的发展,例如在这种分析中纳入蛋白质的三维结构,将改善S-亚硝基化位点的鉴定。