Dasmeh Pouria, Serohijos Adrian W R, Kepp Kasper P, Shakhnovich Eugene I
Department of Chemistry and Chemical Biology, Harvard University DTU Chemistry, Technical University of Denmark, Kongens Lyngby, Denmark Present address: Max Planck Institute of Immunobiology and Epigenetics, Stübeweg, Freiburg, Germany
Department of Chemistry and Chemical Biology, Harvard University
Genome Biol Evol. 2014 Oct 28;6(10):2956-67. doi: 10.1093/gbe/evu223.
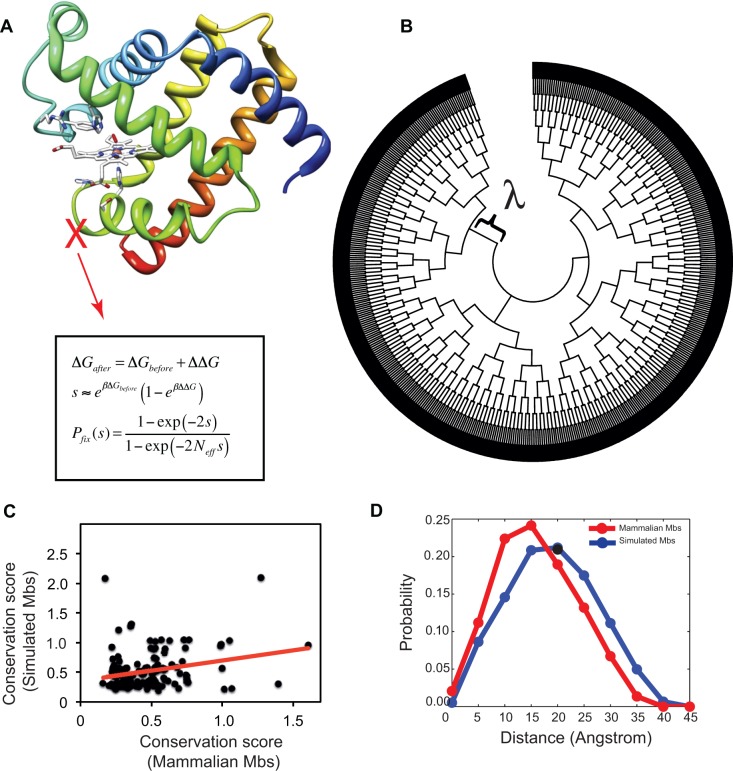
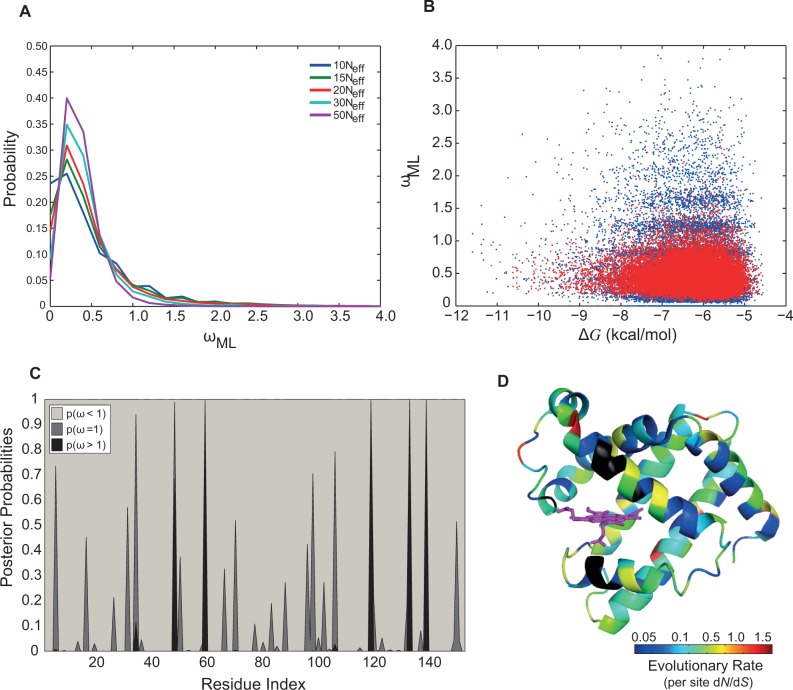
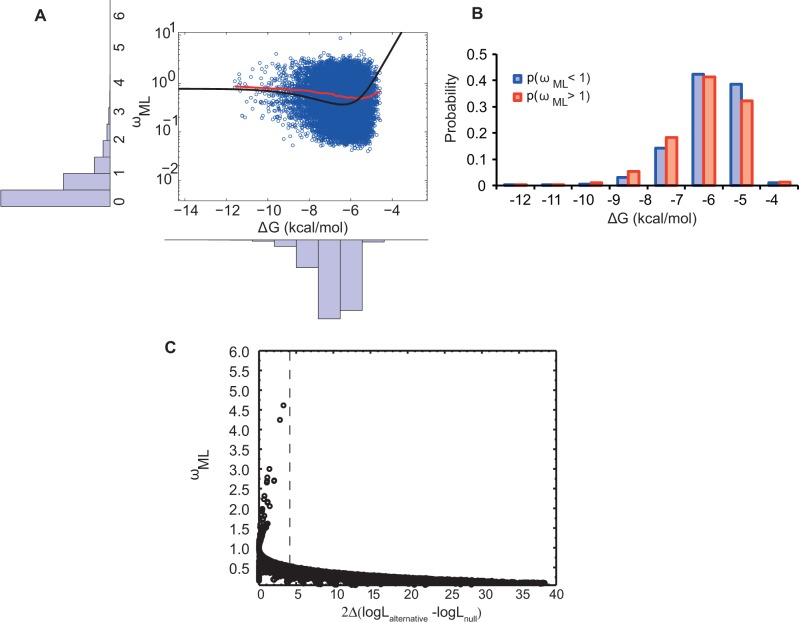
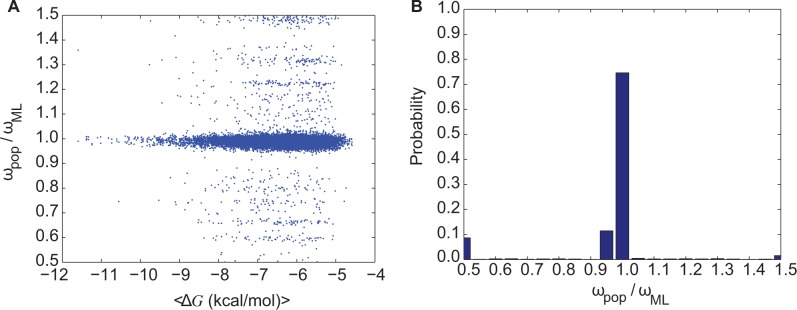
Understanding the relative contributions of various evolutionary processes-purifying selection, neutral drift, and adaptation-is fundamental to evolutionary biology. A common metric to distinguish these processes is the ratio of nonsynonymous to synonymous substitutions (i.e., dN/dS) interpreted from the neutral theory as a null model. However, from biophysical considerations, mutations have non-negligible effects on the biophysical properties of proteins such as folding stability. In this work, we investigated how stability affects the rate of protein evolution in phylogenetic trees by using simulations that combine explicit protein sequences with associated stability changes. We first simulated myoglobin evolution in phylogenetic trees with a biophysically realistic approach that accounts for 3D structural information and estimates of changes in stability upon mutation. We then compared evolutionary rates inferred directly from simulation to those estimated using maximum-likelihood (ML) methods. We found that the dN/dS estimated by ML methods (ωML) is highly predictive of the per gene dN/dS inferred from the simulated phylogenetic trees. This agreement is strong in the regime of high stability where protein evolution is neutral. At low folding stabilities and under mutation-selection balance, we observe deviations from neutrality (per gene dN/dS > 1 and dN/dS < 1). We showed that although per gene dN/dS is robust to these deviations, ML tests for positive selection detect statistically significant per site dN/dS > 1. Altogether, we show how protein biophysics affects the dN/dS estimations and its subsequent interpretation. These results are important for improving the current approaches for detecting positive selection.
理解各种进化过程——纯化选择、中性漂变和适应性——的相对贡献是进化生物学的基础。区分这些过程的一个常用指标是非同义替换与同义替换的比率(即dN/dS),从中性理论的角度将其解释为一个零模型。然而,从生物物理学的角度考虑,突变对蛋白质的生物物理性质(如折叠稳定性)有不可忽略的影响。在这项工作中,我们通过将明确的蛋白质序列与相关的稳定性变化相结合的模拟,研究了稳定性如何影响系统发育树中蛋白质的进化速率。我们首先用一种生物物理上现实的方法模拟了肌红蛋白在系统发育树中的进化,该方法考虑了三维结构信息和突变时稳定性变化的估计。然后,我们将直接从模拟中推断出的进化速率与使用最大似然(ML)方法估计的进化速率进行了比较。我们发现,由ML方法估计的dN/dS(ωML)能够高度预测从模拟系统发育树中推断出的每个基因的dN/dS。在蛋白质进化呈中性的高稳定性状态下,这种一致性很强。在低折叠稳定性和突变-选择平衡的情况下,我们观察到偏离中性的情况(每个基因的dN/dS>1和dN/dS<1)。我们表明,尽管每个基因的dN/dS对这些偏差具有鲁棒性,但用于检测正选择的ML检验检测到每个位点的dN/dS>1具有统计学意义。总之,我们展示了蛋白质生物物理学如何影响dN/dS估计及其后续解释。这些结果对于改进当前检测正选择的方法很重要。