Wang Yuanyuan, Qi Xin, Li Dehai, Zhu Tianjiao, Mo Xiaomei, Li Jing
Key Laboratory of Marine Drugs, Ministry of Education, School of Medicine and Pharmacy, Ocean University of China, Qingdao, People's Republic of China.
Drug Des Devel Ther. 2014 Oct 17;8:1965-77. doi: 10.2147/DDDT.S64989. eCollection 2014.
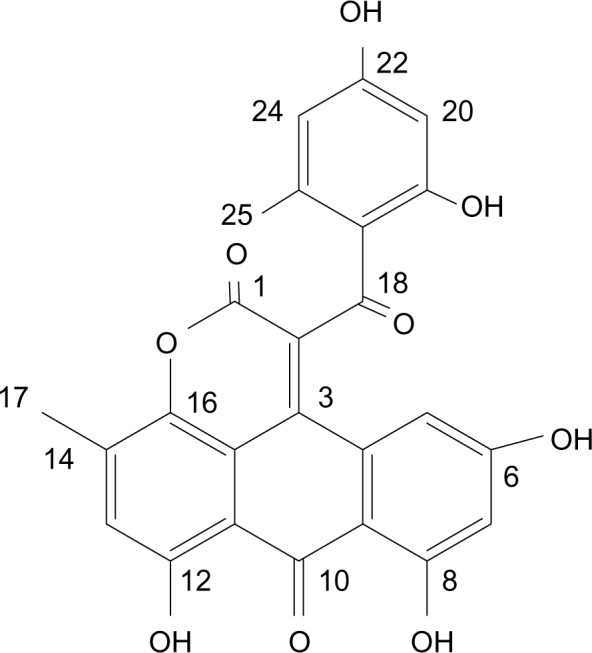
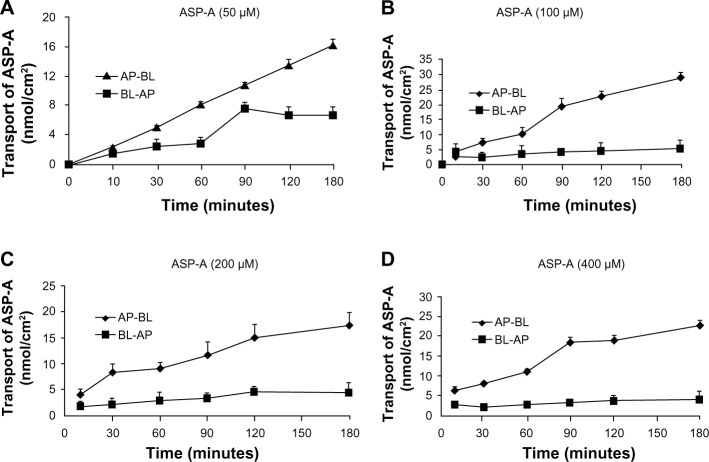
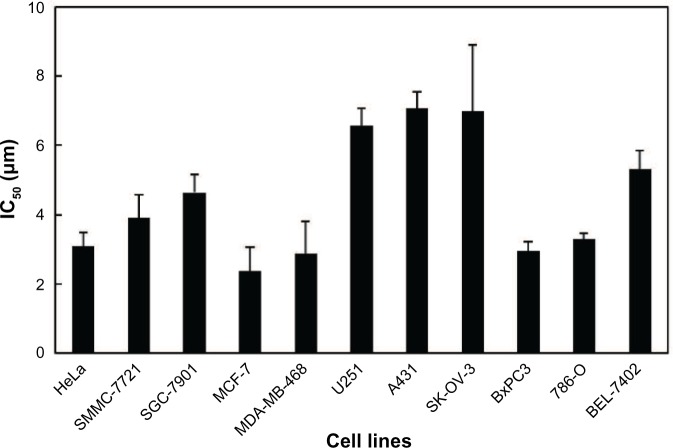
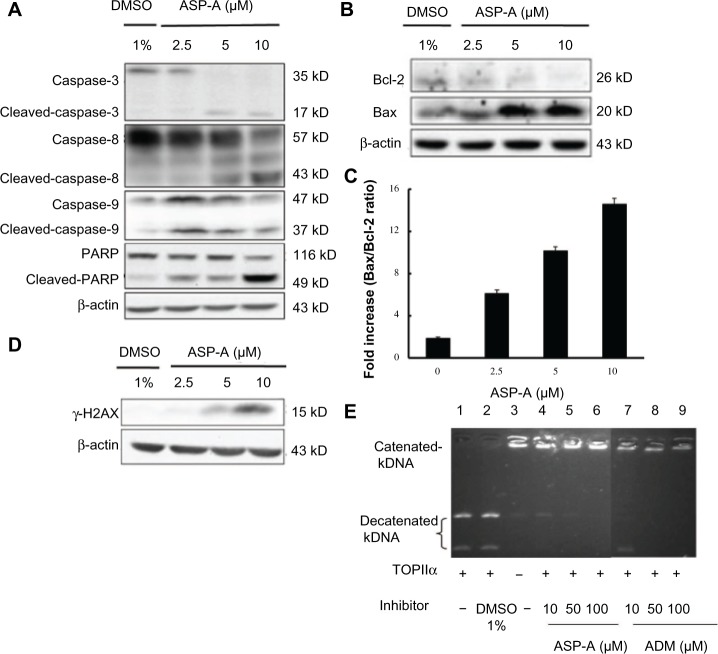
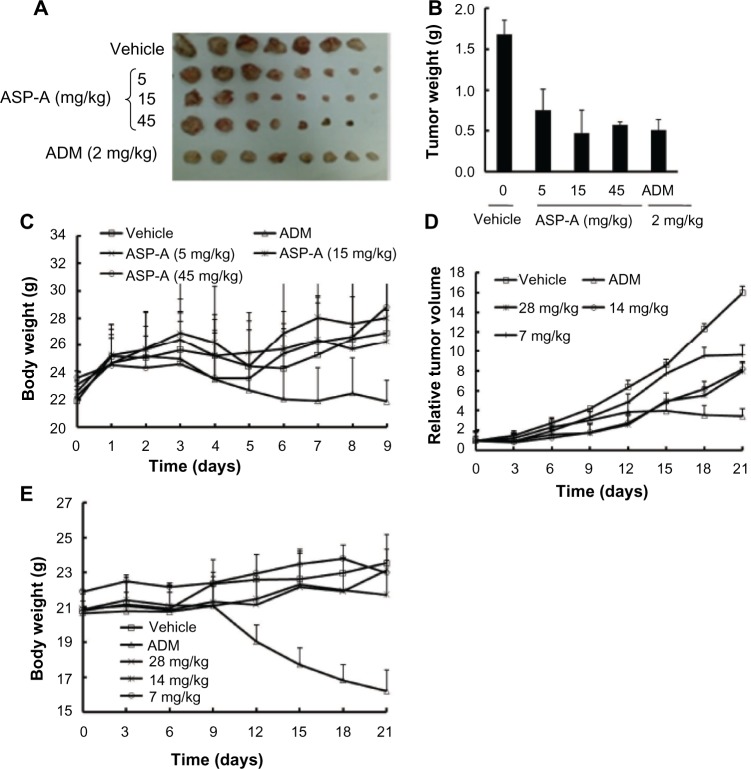
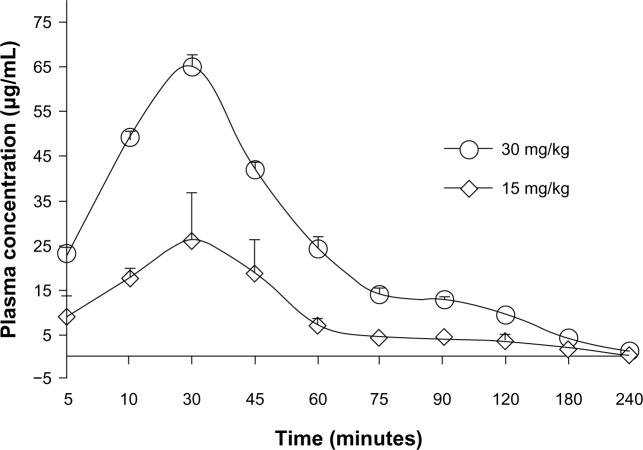
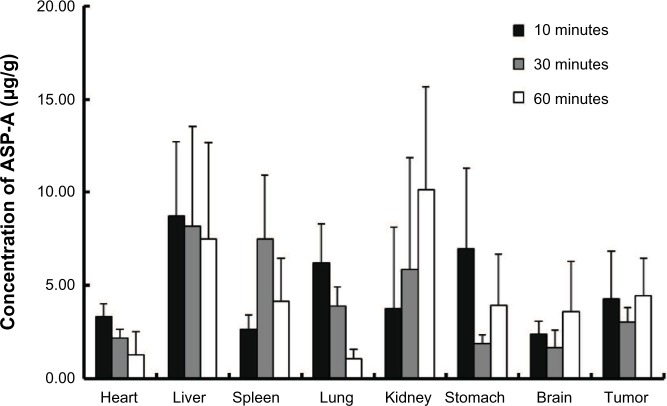
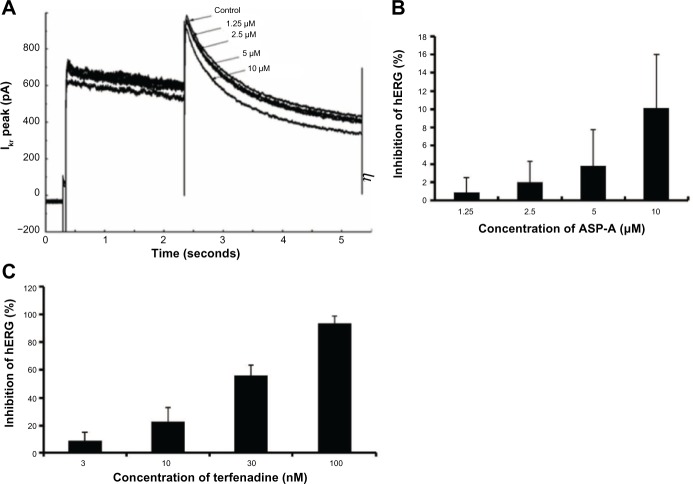
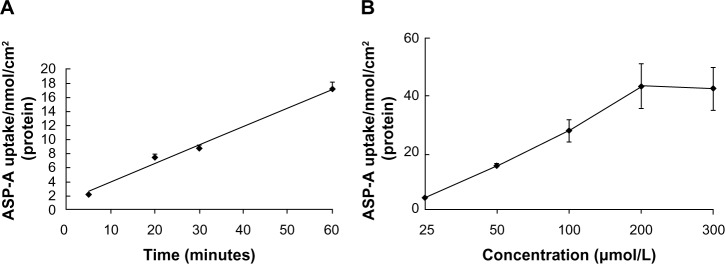
Since the first anthracycline was discovered, many other related compounds have been studied in order to overcome its defects and improve efficacy. In the present paper, we investigated the anticancer effects of a new anthracycline, aspergiolide A (ASP-A), from a marine-derived fungus in vitro and in vivo, and we evaluated the absorption, distribution, metabolism, and toxicity drug properties in early drug development. We found that ASP-A had activity against topoisomerase II that was comparable to adriamycin. ASP-A decreased the growth of various human cancer cells in vitro and induced apoptosis in BEL-7402 cells via a caspase-dependent pathway. The anticancer efficacy of ASP-A on the growth of hepatocellular carcinoma xenografts was further assessed in vivo. Results showed that, compared with the vehicle group, ASP-A exhibited significant anticancer activity with less loss of body weight. A pharmacokinetics and tissue distribution study revealed that ASP-A was rapidly cleared in a first order reaction kinetics manner, and was enriched in cancer tissue. The maximal tolerable dose (MTD) of ASP-A was more than 400 mg/kg, and ASP-A was not considered to be potentially genotoxic or cardiotoxic, as no significant increase of micronucleus rates or inhibition of the hERG channel was seen. Finally, an uptake and transport assay of ASP-A was performed in monolayers of Caco-2 cells, and ASP-A was shown to be absorbed through the active transport pathway. Altogether, these results indicate that ASP-A has anticancer activity targeting topoisomerase II, with a similar structure and mechanism to adriamycin, but with much lower toxicity. Nonetheless, further molecular structure optimization is necessary.
自从第一种蒽环类药物被发现以来,人们对许多其他相关化合物进行了研究,以克服其缺陷并提高疗效。在本文中,我们研究了一种来自海洋真菌的新型蒽环类药物曲霉内酯A(ASP-A)在体外和体内的抗癌作用,并在药物研发早期评估了其吸收、分布、代谢和毒性等药物特性。我们发现ASP-A对拓扑异构酶II具有与阿霉素相当的活性。ASP-A在体外可抑制多种人类癌细胞的生长,并通过半胱天冬酶依赖性途径诱导BEL-7402细胞凋亡。进一步在体内评估了ASP-A对肝癌异种移植瘤生长的抗癌疗效。结果表明,与赋形剂组相比,ASP-A表现出显著的抗癌活性,且体重减轻较少。药代动力学和组织分布研究表明,ASP-A以一级反应动力学方式迅速清除,并在癌组织中富集。ASP-A的最大耐受剂量(MTD)超过400mg/kg,且未观察到微核率显著增加或对hERG通道的抑制,因此不认为ASP-A具有潜在的遗传毒性或心脏毒性。最后,在Caco-2细胞单层中进行了ASP-A的摄取和转运试验,结果表明ASP-A通过主动转运途径被吸收。总之,这些结果表明ASP-A具有靶向拓扑异构酶II的抗癌活性,其结构和作用机制与阿霉素相似,但毒性低得多。尽管如此,仍有必要进一步优化其分子结构。