Lee Donghyung, Williamson Vernell S, Bigdeli T Bernard, Riley Brien P, Fanous Ayman H, Vladimirov Vladimir I, Bacanu Silviu-Alin
Department of Psychiatry, Virginia Institute for Psychiatric and Behavioral Genetics, Center for Biomarker Research & Personalized Medicine, Virginia Commonwealth University, Richmond, VA 23298, USA and Lieber Institute for Brain Development, Johns Hopkins University, Baltimore, MD 21205, USA.
Department of Psychiatry, Virginia Institute for Psychiatric and Behavioral Genetics, Center for Biomarker Research & Personalized Medicine, Virginia Commonwealth University, Richmond, VA 23298, USA and Lieber Institute for Brain Development, Johns Hopkins University, Baltimore, MD 21205, USA Department of Psychiatry, Virginia Institute for Psychiatric and Behavioral Genetics, Center for Biomarker Research & Personalized Medicine, Virginia Commonwealth University, Richmond, VA 23298, USA and Lieber Institute for Brain Development, Johns Hopkins University, Baltimore, MD 21205, USA Department of Psychiatry, Virginia Institute for Psychiatric and Behavioral Genetics, Center for Biomarker Research & Personalized Medicine, Virginia Commonwealth University, Richmond, VA 23298, USA and Lieber Institute for Brain Development, Johns Hopkins University, Baltimore, MD 21205, USA.
Bioinformatics. 2015 Apr 15;31(8):1176-82. doi: 10.1093/bioinformatics/btu816. Epub 2014 Dec 12.
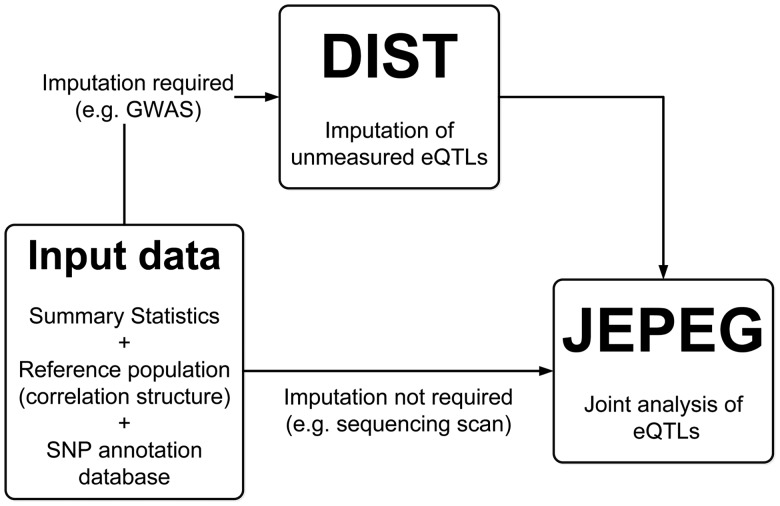
Gene expression is influenced by variants commonly known as expression quantitative trait loci (eQTL). On the basis of this fact, researchers proposed to use eQTL/functional information univariately for prioritizing single nucleotide polymorphisms (SNPs) signals from genome-wide association studies (GWAS). However, most genes are influenced by multiple eQTLs which, thus, jointly affect any downstream phenotype. Therefore, when compared with the univariate prioritization approach, a joint modeling of eQTL action on phenotypes has the potential to substantially increase signal detection power. Nonetheless, a joint eQTL analysis is impeded by (i) not measuring all eQTLs in a gene and/or (ii) lack of access to individual genotypes.
We propose joint effect on phenotype of eQTL/functional SNPs associated with a gene (JEPEG), a novel software tool which uses only GWAS summary statistics to (i) impute the summary statistics at unmeasured eQTLs and (ii) test for the joint effect of all measured and imputed eQTLs in a gene. We illustrate the behavior/performance of the developed tool by analysing the GWAS meta-analysis summary statistics from the Psychiatric Genomics Consortium Stage 1 and the Genetic Consortium for Anorexia Nervosa.
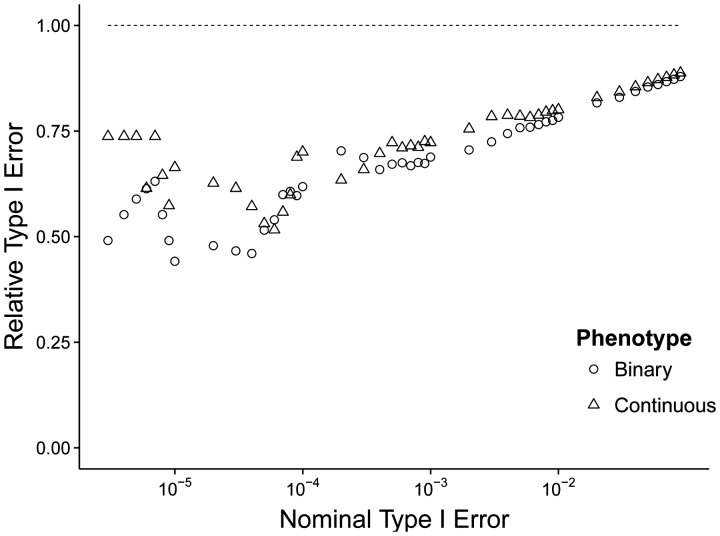
Applied analyses results suggest that JEPEG complements commonly used univariate GWAS tools by: (i) increasing signal detection power via uncovering (a) novel genes or (b) known associated genes in smaller cohorts and (ii) assisting in fine-mapping of challenging regions, e.g. major histocompatibility complex for schizophrenia.
JEPEG, its associated database of eQTL SNPs and usage examples are publicly available at http://code.google.com/p/jepeg/.
Supplementary data are available at Bioinformatics online.
基因表达受通常称为表达数量性状位点(eQTL)的变异影响。基于这一事实,研究人员提议单独使用eQTL/功能信息对全基因组关联研究(GWAS)中的单核苷酸多态性(SNP)信号进行优先级排序。然而,大多数基因受多个eQTL影响,因此这些eQTL共同影响任何下游表型。因此,与单变量优先级排序方法相比,对eQTL对表型的作用进行联合建模有可能大幅提高信号检测能力。尽管如此,联合eQTL分析受到以下因素的阻碍:(i)未测量基因中的所有eQTL和/或(ii)无法获取个体基因型。
我们提出了与基因相关的eQTL/功能SNP对表型的联合效应(JEPEG),这是一种新颖的软件工具,它仅使用GWAS汇总统计信息来(i)估算未测量eQTL处的汇总统计信息,以及(ii)测试基因中所有测量和估算的eQTL的联合效应。我们通过分析精神病基因组学联盟第一阶段的GWAS荟萃分析汇总统计信息和神经性厌食症遗传联盟的数据来说明所开发工具的行为/性能。
应用分析结果表明,JEPEG通过以下方式补充了常用的单变量GWAS工具:(i)通过在较小队列中发现(a)新基因或(b)已知相关基因来提高信号检测能力,以及(ii)协助对具有挑战性的区域进行精细定位,例如精神分裂症的主要组织相容性复合体。
JEPEG及其相关的eQTL SNP数据库和使用示例可在http://code.google.com/p/jepeg/上公开获取。
补充数据可在《生物信息学》在线版获取。