Wang Heming, Zhu Xiaofeng
Department of Epidemiology and Biostatistics, Case Western Reserve University, 10900 Euclid Ave, Cleveland, OH 44106-4945, USA.
BMC Proc. 2014 Jun 17;8(Suppl 1 Genetic Analysis Workshop 18Vanessa Olmo):S24. doi: 10.1186/1753-6561-8-S1-S24. eCollection 2014.
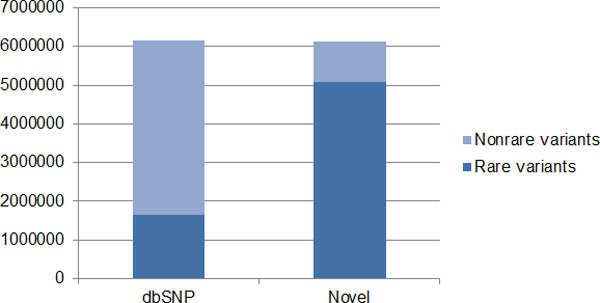
De novo mutations enrich the sequence diversity and carry the clue of evolutional selection. Recent studies suggest the de novo mutations could be one of the risk factors for complex diseases. We conducted a survey of de novo mutations using the whole genome sequence data but only available on the odd autosomes of Mexican American families provided by Genetic Analysis Workshop 18. We extracted 8 three-generation families who have sequencing data available from 20 large pedigrees. By comparing the known single nucleotide variants (SNVs) in dbSNP129 and the de novo variants transmitted in the Mexican American families, we were able to estimate a de novo mutation rate of 1.64(±0.42) × 10(-8) per position per haploid genome. This result is consistent with the estimates in literature that required many extensive validation efforts, such as genotyping and further resequencing. Our analysis suggests the importance of using family samples for studying rare variants.
新生突变丰富了序列多样性,并承载着进化选择的线索。近期研究表明,新生突变可能是复杂疾病的风险因素之一。我们利用全基因组序列数据对新生突变进行了一项调查,但数据仅来自遗传分析研讨会18提供的墨西哥裔美国家庭的奇数常染色体。我们从20个大家系中提取了8个有测序数据的三代家庭。通过比较dbSNP129中已知的单核苷酸变异(SNV)和墨西哥裔美国家庭中传递的新生变异,我们能够估计出每个单倍体基因组每个位置的新生突变率为1.64(±0.42)×10^(-8)。这一结果与文献中的估计一致,而这些估计需要许多广泛的验证工作,如基因分型和进一步的重测序。我们的分析表明了使用家系样本研究罕见变异的重要性。