Inokuchi Ryota, Kurata Hideaki, Endo Kiyoshi, Kitsuta Yoichi, Nakajima Susumu, Hatamochi Atsushi, Yahagi Naoki
From the Department of Emergency and Critical Care Medicine, The University of Tokyo Hospital, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8655, Japan (RI, HK, YK, SN, NY); Department of Emergency Medicine, JR General Hospital, 2-1-3 Yoyogi, Shibuya-ku, Tokyo 151-8528, Japan (RI, KE); Department of Surgery, Hanna Central Hospital, 741 Tawaraguchi, Ikoma city, Nara 630-0243, Japan (HK); and Department of Dermatology, Dokkyo Medical University, School of Medicine, Kitakobayashi, Mibu, Tochigi 321-0293, Japan (AH).
Medicine (Baltimore). 2014 Dec;93(28):e291. doi: 10.1097/MD.0000000000000291.
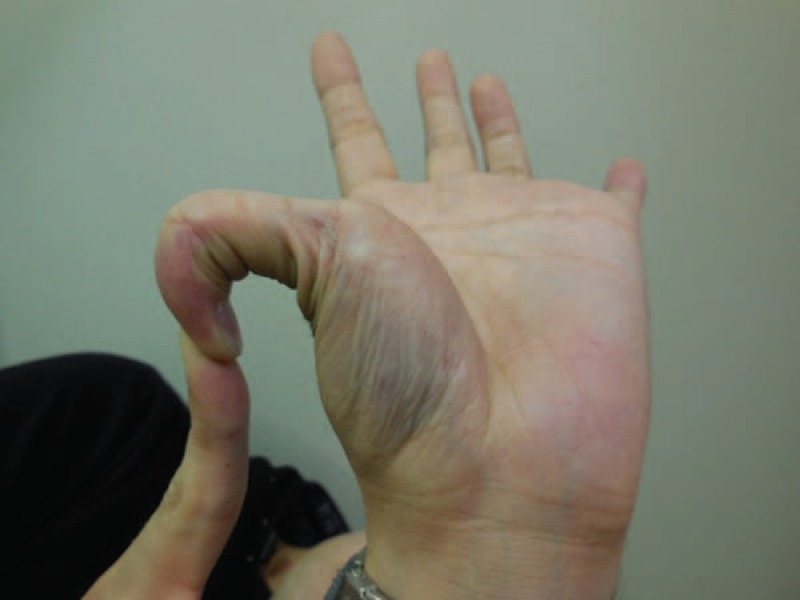
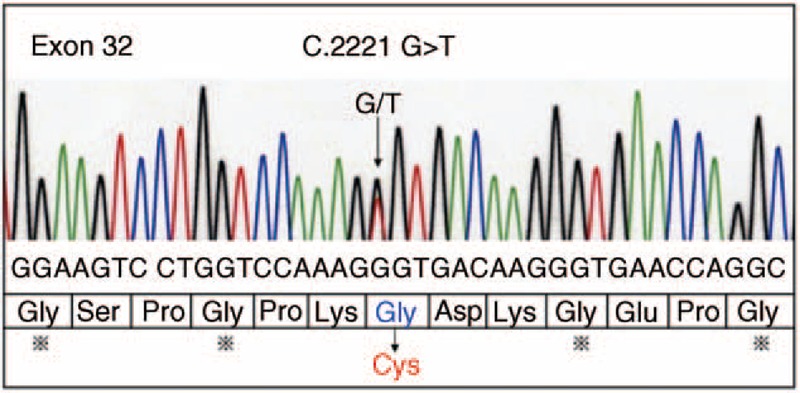
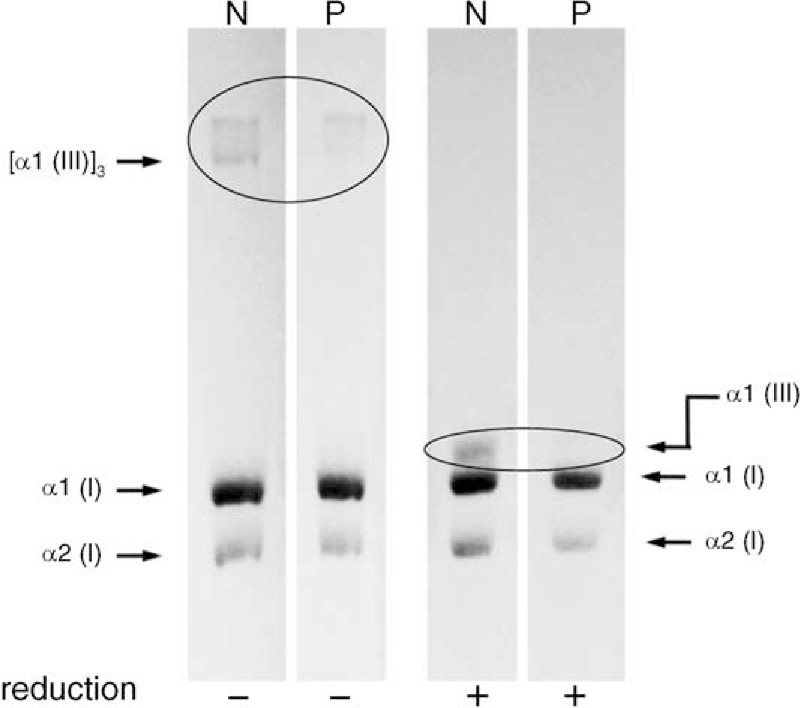
As a type of Ehlers-Danlos syndrome (EDS), vascular EDs (vEDS) is typified by a number of characteristic facial features (eg, large eyes, small chin, sunken cheeks, thin nose and lips, lobeless ears). However, vEDs does not typically display hypermobility of the large joints and skin hyperextensibility, which are features typical of the more common forms of EDS. Thus, colonic perforation or aneurysm rupture may be the first presentation of the disease. Because both complications are associated with a reduced life expectancy for individuals with this condition, an awareness of the clinical features of vEDS is important. Here, we describe the treatment of vEDS lacking the characteristic facial attributes in a 24-year-old healthy man who presented to the emergency room with abdominal pain. Enhanced computed tomography revealed diverticula and perforation in the sigmoid colon. The lesion of the sigmoid colon perforation was removed, and Hartmann procedure was performed. During the surgery, the control of bleeding was required because of vascular fragility. Subsequent molecular and genetic analysis was performed based on the suspected diagnosis of vEDS. These analyses revealed reduced type III collagen synthesis in cultured skin fibroblasts and identified a previously undocumented mutation in the gene for a1 type III collagen, confirming the diagnosis of vEDS. After eliciting a detailed medical profile, we learned his mother had a history of extensive bruising since childhood and idiopathic hematothorax. Both were prescribed oral celiprolol. One year after admission, the patient was free of recurrent perforation. This case illustrates an awareness of the clinical characteristics of vEDS and the family history is important because of the high mortality from this condition even in young people. Importantly, genetic assays could help in determining the surgical procedure and offer benefits to relatives since this condition is inherited in an autosomal dominant manner.
作为埃勒斯-当洛综合征(EDS)的一种类型,血管型EDS(vEDS)具有一些特征性面部特征(如大眼睛、小下巴、脸颊凹陷、薄鼻和嘴唇、无耳垂)。然而,vEDS通常不表现出大关节的活动过度和皮肤过度伸展,而这些是更常见的EDS形式的典型特征。因此,结肠穿孔或动脉瘤破裂可能是该疾病的首发表现。由于这两种并发症都与患有这种疾病的个体预期寿命缩短有关,了解vEDS的临床特征很重要。在此,我们描述了一名24岁健康男性因腹痛就诊于急诊室,其vEDS缺乏特征性面部特征的治疗情况。增强计算机断层扫描显示乙状结肠憩室和穿孔。切除乙状结肠穿孔病变,并进行了哈特曼手术。手术过程中,由于血管脆弱需要控制出血。随后基于疑似vEDS的诊断进行了分子和基因分析。这些分析显示培养的皮肤成纤维细胞中III型胶原蛋白合成减少,并在α1 III型胶原蛋白基因中鉴定出一个以前未记录的突变,从而确诊为vEDS。在了解详细的病史后,我们得知他的母亲自童年起就有广泛瘀伤史和特发性血胸史。两人都服用了口服塞利洛尔。入院一年后,患者未再发生穿孔。该病例说明了解vEDS的临床特征以及家族史很重要,因为即使在年轻人中这种疾病的死亡率也很高。重要的是,基因检测有助于确定手术方案,并对亲属有益,因为这种疾病是以常染色体显性方式遗传的。