González-del Pozo María, Méndez-Vidal Cristina, Bravo-Gil Nereida, Vela-Boza Alicia, Dopazo Joaquin, Borrego Salud, Antiñolo Guillermo
Department of Genetics, Reproduction and Fetal Medicine, Institute of Biomedicine of Seville, University Hospital Virgen del Rocío/CSIC/University of Seville, Seville, Spain; Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER), Seville, Spain.
Genomics and Bioinformatics Platform of Andalusia (GBPA), Seville, Spain.
PLoS One. 2014 Dec 29;9(12):e116176. doi: 10.1371/journal.pone.0116176. eCollection 2014.
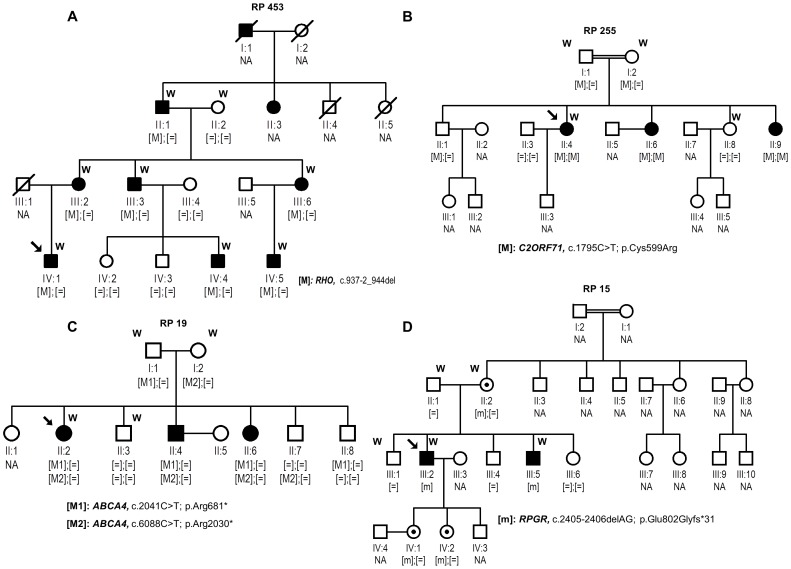
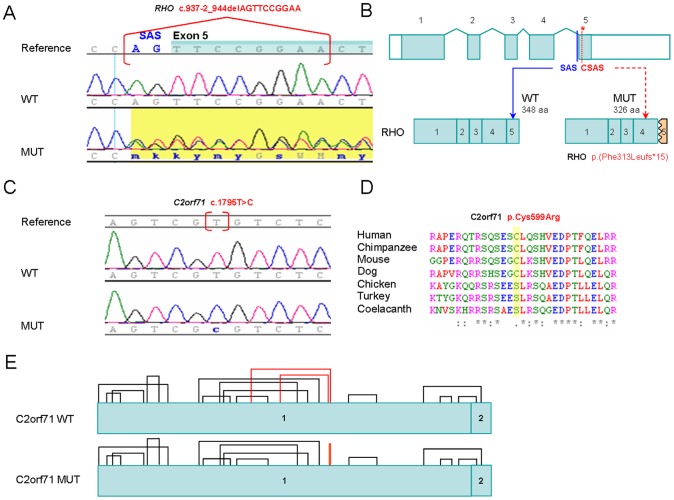
This study aimed to identify the underlying molecular genetic cause in four Spanish families clinically diagnosed of Retinitis Pigmentosa (RP), comprising one autosomal dominant RP (adRP), two autosomal recessive RP (arRP) and one with two possible modes of inheritance: arRP or X-Linked RP (XLRP). We performed whole exome sequencing (WES) using NimbleGen SeqCap EZ Exome V3 sample preparation kit and SOLID 5500xl platform. All variants passing filter criteria were validated by Sanger sequencing to confirm familial segregation and the absence in local control population. This strategy allowed the detection of: (i) one novel heterozygous splice-site deletion in RHO, c.937-2_944del, (ii) one rare homozygous mutation in C2orf71, c.1795T>C; p.Cys599Arg, not previously associated with the disease, (iii) two heterozygous null mutations in ABCA4, c.2041C>T; p.R681* and c.6088C>T; p.R2030*, and (iv) one mutation, c.2405-2406delAG; p.Glu802Glyfs*31 in the ORF15 of RPGR. The molecular findings for RHO and C2orf71 confirmed the initial diagnosis of adRP and arRP, respectively, while patients with the two ABCA4 mutations, both previously associated with Stargardt disease, presented symptoms of RP with early macular involvement. Finally, the X-Linked inheritance was confirmed for the family with the RPGR mutation. This latter finding allowed the inclusion of carrier sisters in our preimplantational genetic diagnosis program.
本研究旨在确定四个临床诊断为色素性视网膜炎(RP)的西班牙家庭潜在的分子遗传病因,其中包括一个常染色体显性RP(adRP)家庭、两个常染色体隐性RP(arRP)家庭以及一个具有两种可能遗传模式的家庭:arRP或X连锁RP(XLRP)。我们使用NimbleGen SeqCap EZ Exome V3样本制备试剂盒和SOLID 5500xl平台进行了全外显子组测序(WES)。所有通过筛选标准的变异均通过桑格测序进行验证,以确认家族分离情况以及在当地对照人群中不存在该变异。该策略检测到:(i)RHO基因中的一个新型杂合剪接位点缺失,c.937 - 2_944del;(ii)C2orf71基因中的一个罕见纯合突变,c.1795T>C;p.Cys599Arg,此前未发现与该疾病相关;(iii)ABCA4基因中的两个杂合无效突变,c.2041C>T;p.R681和c.6088C>T;p.R2030;以及(iv)RPGR基因ORF15中的一个突变,c.2405 - 2406delAG;p.Glu802Glyfs*31。RHO和C2orf71的分子检测结果分别证实了最初诊断的adRP和arRP,而两个携带ABCA4突变的患者,这两个突变此前均与斯塔加特病相关,表现出RP症状且早期黄斑受累。最后确诊携带RPGR突变的家庭为X连锁遗传。这一发现使得我们能够将携带者姐妹纳入植入前基因诊断项目。