Eyal Eran, Lum Gengkon, Bahar Ivet
Cancer Research Institute, Sheba Medical Center, 2 Sheba Rd, Ramat Gan 52621, Israel and Department of Computational and System Biology, University of Pittsburgh, 3501 Fifth Ave, Pittsburgh, PA 15213, USA.
Bioinformatics. 2015 May 1;31(9):1487-9. doi: 10.1093/bioinformatics/btu847. Epub 2015 Jan 6.
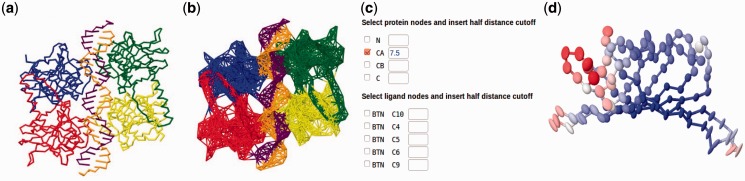
The anisotropic network model (ANM) is one of the simplest yet powerful tools for exploring protein dynamics. Its main utility is to predict and visualize the collective motions of large complexes and assemblies near their equilibrium structures. The ANM server, introduced by us in 2006 helped making this tool more accessible to non-sophisticated users. We now provide a new version (ANM 2.0), which allows inclusion of nucleic acids and ligands in the network model and thus enables the investigation of the collective motions of protein-DNA/RNA and -ligand systems. The new version offers the flexibility of defining the system nodes and the interaction types and cutoffs. It also includes extensive improvements in hardware, software and graphical interfaces.
ANM 2.0 is available at http://anm.csb.pitt.edu
各向异性网络模型(ANM)是探索蛋白质动力学最简单却最强大的工具之一。其主要用途是预测和可视化大型复合物及组件在其平衡结构附近的集体运动。我们于2006年推出的ANM服务器,有助于让非专业用户更易使用该工具。我们现在提供了一个新版本(ANM 2.0),它允许在网络模型中纳入核酸和配体,从而能够研究蛋白质 - DNA/RNA和 - 配体系统的集体运动。新版本提供了定义系统节点、相互作用类型和截止值的灵活性。它还在硬件、软件和图形界面方面有了广泛改进。
ANM 2.0可在http://anm.csb.pitt.edu获取。