Stetson Lindsay C, Pearl Taylor, Chen Yanwen, Barnholtz-Sloan Jill S
BMC Genomics. 2014;15 Suppl 7(Suppl 7):S2. doi: 10.1186/1471-2164-15-S7-S2. Epub 2014 Oct 27.
A challenge in precision medicine is the transformation of genomic data into knowledge that can be used to stratify patients into treatment groups based on predicted clinical response. Although clinical trials remain the only way to truly measure drug toxicities and effectiveness, as a scientific community we lack the resources to clinically assess all drugs presently under development. Therefore, an effective preclinical model system that enables prediction of anticancer drug response could significantly speed the broader adoption of personalized medicine.
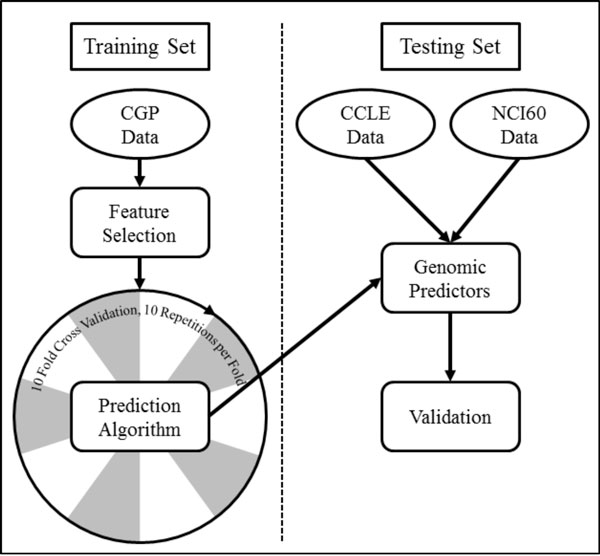
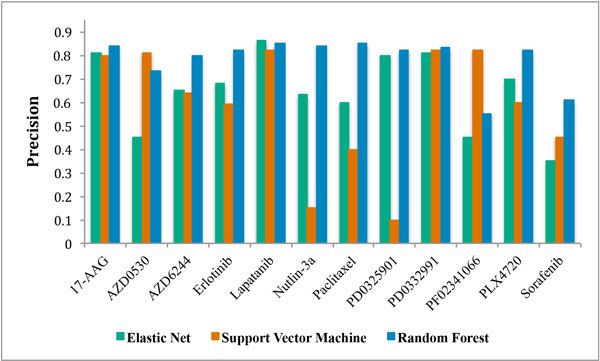
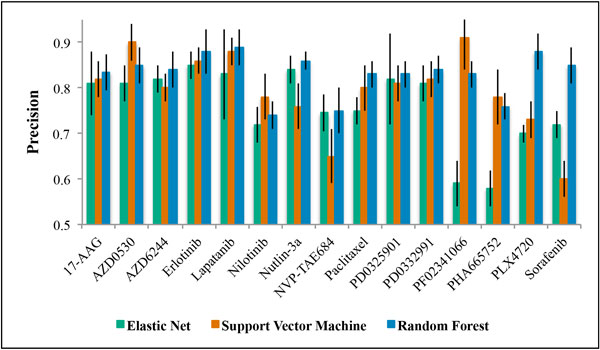
Three large-scale pharmacogenomic studies have screened anticancer compounds in greater than 1000 distinct human cancer cell lines. We combined these datasets to generate and validate multi-omic predictors of drug response. We compared drug response signatures built using a penalized linear regression model and two non-linear machine learning techniques, random forest and support vector machine. The precision and robustness of each drug response signature was assessed using cross-validation across three independent datasets. Fifteen drugs were common among the datasets. We validated prediction signatures for eleven out of fifteen tested drugs (17-AAG, AZD0530, AZD6244, Erlotinib, Lapatinib, Nultin-3, Paclitaxel, PD0325901, PD0332991, PF02341066, and PLX4720).
Multi-omic predictors of drug response can be generated and validated for many drugs. Specifically, the random forest algorithm generated more precise and robust prediction signatures when compared to support vector machines and the more commonly used elastic net regression. The resulting drug response signatures can be used to stratify patients into treatment groups based on their individual tumor biology, with two major benefits: speeding the process of bringing preclinical drugs to market, and the repurposing and repositioning of existing anticancer therapies.
精准医学面临的一个挑战是将基因组数据转化为可用于根据预测的临床反应将患者分层到治疗组的知识。尽管临床试验仍然是真正衡量药物毒性和有效性的唯一方法,但作为一个科学界,我们缺乏资源对目前正在研发的所有药物进行临床评估。因此,一个能够预测抗癌药物反应的有效的临床前模型系统可以显著加速个性化医学的更广泛应用。
三项大规模药物基因组学研究在1000多种不同的人类癌细胞系中筛选了抗癌化合物。我们合并了这些数据集以生成和验证药物反应的多组学预测因子。我们比较了使用惩罚线性回归模型以及两种非线性机器学习技术(随机森林和支持向量机)构建的药物反应特征。使用三个独立数据集的交叉验证评估了每个药物反应特征的准确性和稳健性。这些数据集中有15种药物是共有的。我们验证了15种测试药物中的11种(17-AAG、AZD0530、AZD6244、厄洛替尼、拉帕替尼、Nultin-3、紫杉醇、PD0325901、PD0332991、PF02341066和PLX4720)的预测特征。
可以针对多种药物生成和验证药物反应的多组学预测因子。具体而言,与支持向量机和更常用的弹性网回归相比,随机森林算法生成的预测特征更精确、更稳健。由此产生的药物反应特征可用于根据患者个体的肿瘤生物学特征将其分层到治疗组,有两个主要好处:加速临床前药物上市的进程,以及现有抗癌疗法的重新利用和重新定位。