Arenas Miguel, Lopes Joao S, Beaumont Mark A, Posada David
Centre for Molecular Biology "Severo Ochoa," Consejo Superior de Investigaciones Científicas (CSIC), Madrid, Spain Departamento de Bioquímica, Genética e Inmunología, Universidad de Vigo, Vigo, Spain
Instituto Gulbenkian de Ciencia, Oeiras, Portugal.
Mol Biol Evol. 2015 Apr;32(4):1109-12. doi: 10.1093/molbev/msu411. Epub 2015 Jan 9.
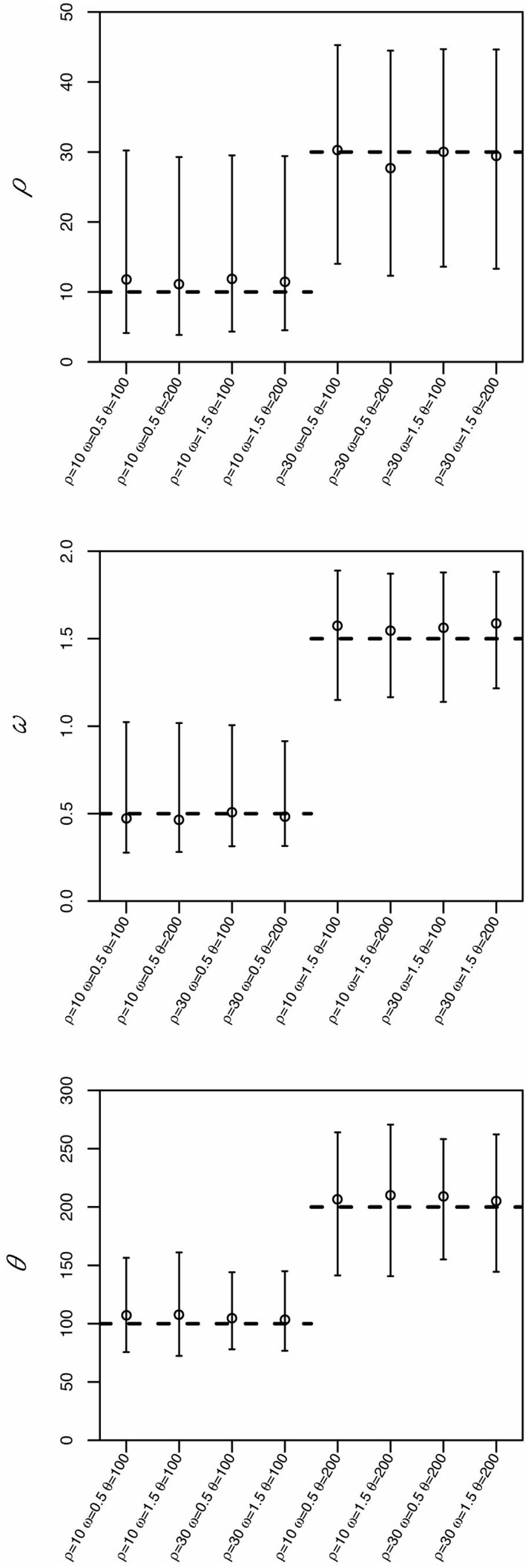
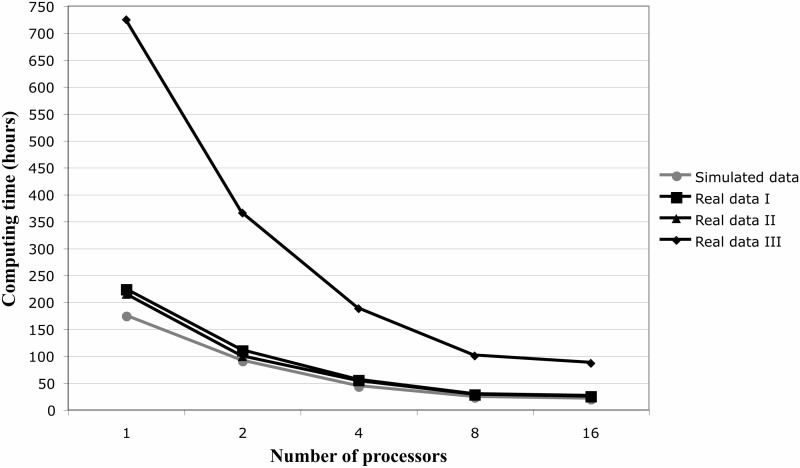
The estimation of substitution and recombination rates can provide important insights into the molecular evolution of protein-coding sequences. Here, we present a new computational framework, called "CodABC," to jointly estimate recombination, substitution and synonymous and nonsynonymous rates from coding data. CodABC uses approximate Bayesian computation with and without regression adjustment and implements a variety of codon models, intracodon recombination, and longitudinal sampling. CodABC can provide accurate joint parameter estimates from recombining coding sequences, often outperforming maximum-likelihood methods based on more approximate models. In addition, CodABC allows for the inclusion of several nuisance parameters such as those representing codon frequencies, transition matrices, heterogeneity across sites or invariable sites. CodABC is freely available from http://code.google.com/p/codabc/, includes a GUI, extensive documentation and ready-to-use examples, and can run in parallel on multicore machines.
替换率和重组率的估计能够为蛋白质编码序列的分子进化提供重要见解。在此,我们提出了一个名为“CodABC”的新计算框架,用于从编码数据中联合估计重组率、替换率以及同义率和非同义率。CodABC使用带有和不带有回归调整的近似贝叶斯计算,并实现了多种密码子模型、密码子内重组以及纵向抽样。CodABC能够从重组的编码序列中提供准确的联合参数估计,通常优于基于更近似模型的最大似然方法。此外,CodABC允许纳入几个干扰参数,例如代表密码子频率、转换矩阵、位点间异质性或不变位点的参数。CodABC可从http://code.google.com/p/codabc/免费获取,包括一个图形用户界面、详尽的文档以及现成可用的示例,并且能够在多核机器上并行运行。