Guo Yunfei, Ding Xiaolei, Shen Yufeng, Lyon Gholson J, Wang Kai
Zilkha Neurogenetic Institute, University of Southern California, Los Angeles, CA 90033, USA.
Department of Preventive Medicine, Keck School of Medicine, University of Southern California, Los Angeles, CA 90032, USA.
Sci Rep. 2015 Sep 18;5:14283. doi: 10.1038/srep14283.
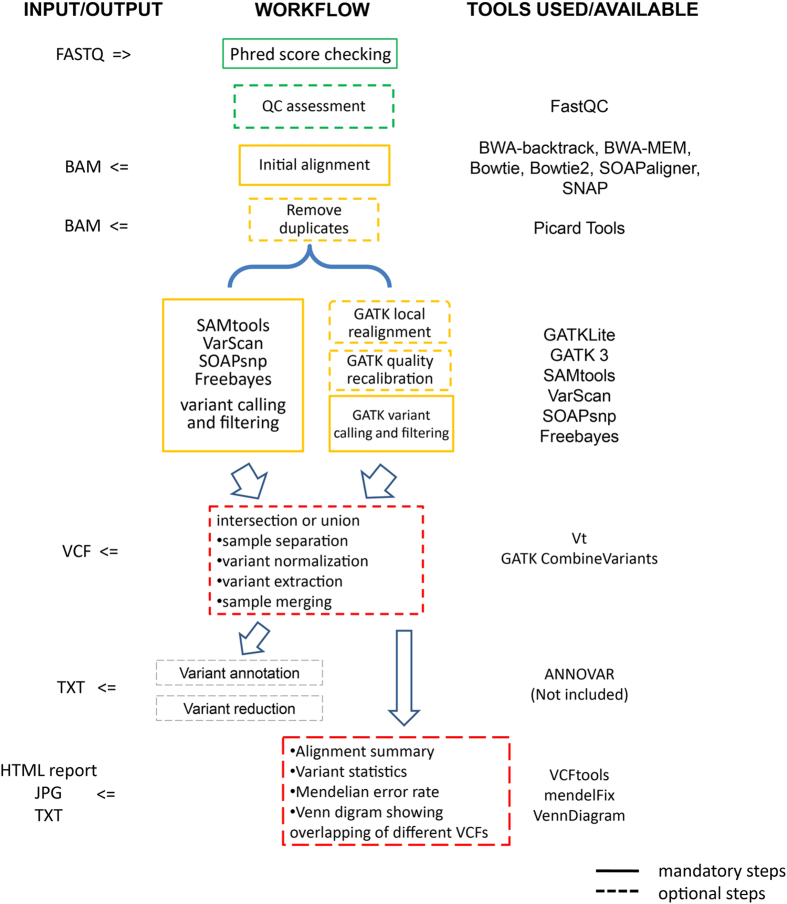
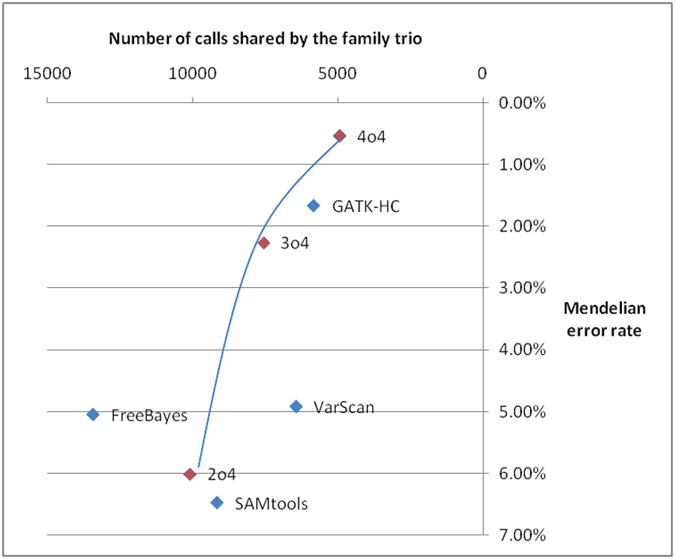
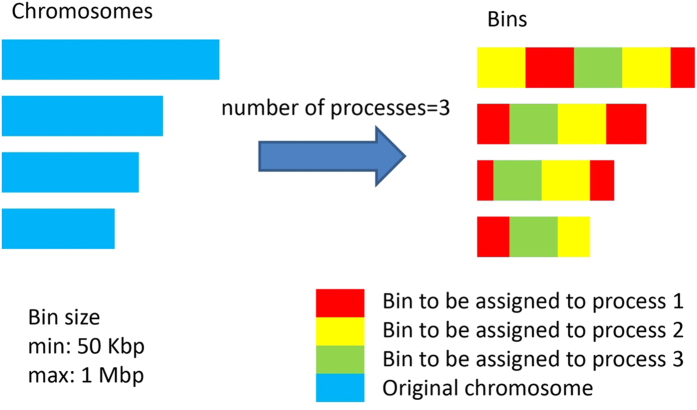
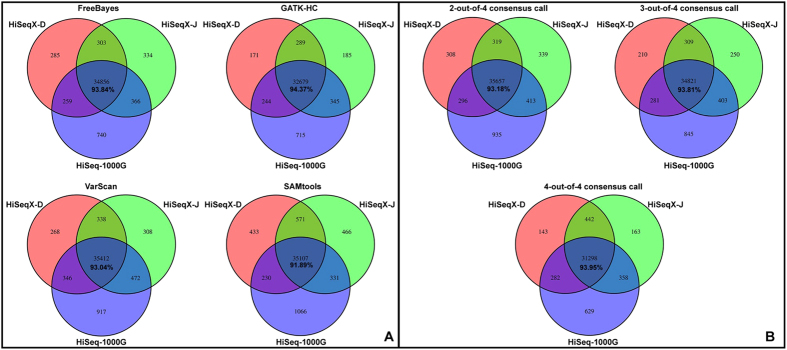
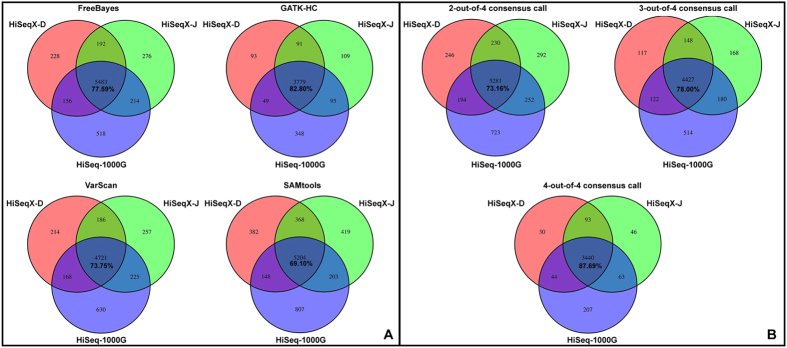
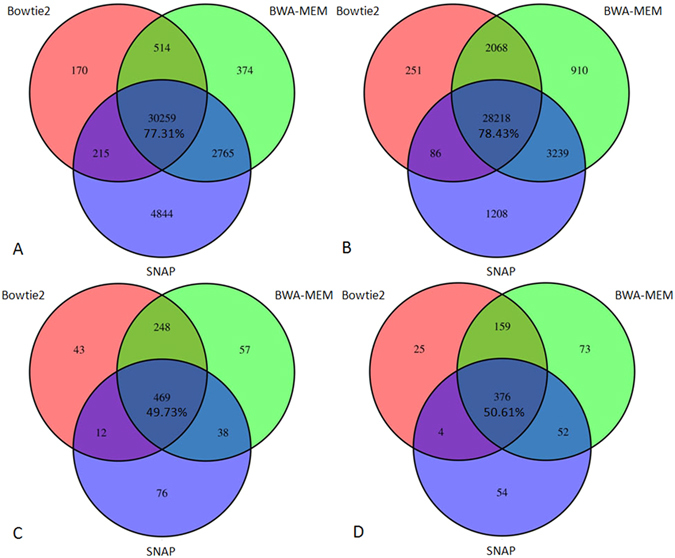
Next-generation sequencing (NGS) technology has greatly helped us identify disease-contributory variants for Mendelian diseases. However, users are often faced with issues such as software compatibility, complicated configuration, and no access to high-performance computing facility. Discrepancies exist among aligners and variant callers. We developed a computational pipeline, SeqMule, to perform automated variant calling from NGS data on human genomes and exomes. SeqMule integrates computational-cluster-free parallelization capability built on top of the variant callers, and facilitates normalization/intersection of variant calls to generate consensus set with high confidence. SeqMule integrates 5 alignment tools, 5 variant calling algorithms and accepts various combinations all by one-line command, therefore allowing highly flexible yet fully automated variant calling. In a modern machine (2 Intel Xeon X5650 CPUs, 48 GB memory), when fast turn-around is needed, SeqMule generates annotated VCF files in a day from a 30X whole-genome sequencing data set; when more accurate calling is needed, SeqMule generates consensus call set that improves over single callers, as measured by both Mendelian error rate and consistency. SeqMule supports Sun Grid Engine for parallel processing, offers turn-key solution for deployment on Amazon Web Services, allows quality check, Mendelian error check, consistency evaluation, HTML-based reports. SeqMule is available at http://seqmule.openbioinformatics.org.
下一代测序(NGS)技术极大地帮助我们识别孟德尔疾病的致病变异。然而,用户经常面临软件兼容性、配置复杂以及无法使用高性能计算设备等问题。比对器和变异检测工具之间存在差异。我们开发了一个计算流程SeqMule,用于对人类基因组和外显子组的NGS数据进行自动变异检测。SeqMule整合了基于变异检测工具的无计算集群并行化能力,并促进变异检测的标准化/交叉分析,以生成高可信度的一致性集合。SeqMule整合了5种比对工具、5种变异检测算法,并且通过一行命令接受各种组合,因此允许高度灵活但完全自动化的变异检测。在一台现代机器(2个英特尔至强X5650 CPU,48GB内存)上,当需要快速周转时,SeqMule能在一天内从30X全基因组测序数据集中生成注释的VCF文件;当需要更准确的检测时,SeqMule生成的一致性检测集合在孟德尔错误率和一致性方面均优于单个检测工具。SeqMule支持使用Sun Grid Engine进行并行处理,为在亚马逊网络服务上的部署提供一站式解决方案,允许进行质量检查、孟德尔错误检查、一致性评估以及基于HTML的报告。SeqMule可在http://seqmule.openbioinformatics.org获取。