Książkiewicz Michał, Zielezinski Andrzej, Wyrwa Katarzyna, Szczepaniak Anna, Rychel Sandra, Karlowski Wojciech, Wolko Bogdan, Naganowska Barbara
Department of Genomics, Institute of Plant Genetics of the Polish Academy of Sciences, Strzeszyńska 34, 60-479 Poznan, Poland.
Institute of Molecular Biology and Biotechnology, Adam Mickiewicz University, Umultowska 89, 61-614 Poznan, Poland.
Plant Mol Biol Report. 2015;33(1):84-101. doi: 10.1007/s11105-014-0730-4.
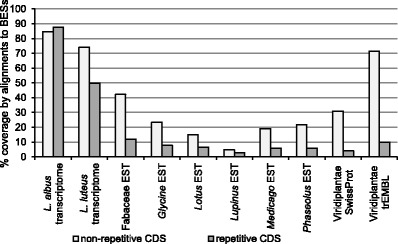
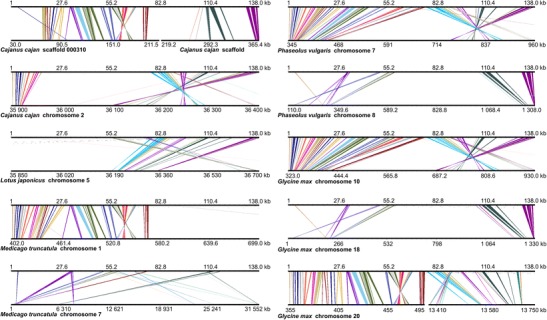
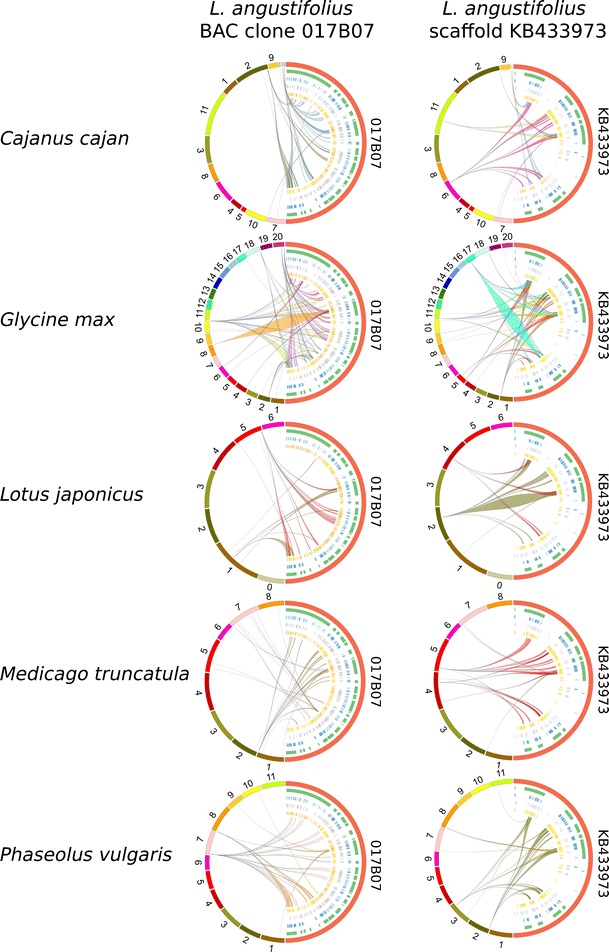
The narrow-leafed lupin () was recently considered as a legume reference species. Genetic resources have been developed, including a draft genome sequence, linkage maps, nuclear DNA libraries, and cytogenetic chromosome-specific landmarks. Here, we used a complex approach, involving DNA fingerprinting, sequencing, genetic mapping, and molecular cytogenetics, to localize and analyze gene-rich regions (GRRs). A genomic bacterial artificial chromosome (BAC) library was screened with short sequence repeat (SSR)-based probes. Selected BACs were fingerprinted and assembled into contigs. BAC-end sequence (BES) annotation allowed us to choose clones for sequencing, targeting GRRs. Additionally, BESs were aligned to the scaffolds of the genome sequence. The genetic map was supplemented with 35 BES-derived markers, distributed in 14 linkage groups and tagging 37 scaffolds. The identified GRRs had an average gene density of 19.6 genes/100 kb and physical-to-genetic distance ratios of 11 to 109 kb/cM. Physical and genetic mapping was supported by multi-BAC-fluorescence in situ hybridization (FISH), and five new linkage groups were assigned to the chromosomes. Syntenic links to the genome sequences of five legume species (, , , , and ) were identified. The comparative mapping of the two largest lupin GRRs provides novel evidence for ancient duplications in all of the studied species. These regions are conserved among representatives of the main clades of Papilionoideae. Furthermore, despite the complex evolution of legumes, some segments of the nuclear genome were not substantially modified and retained their quasi-ancestral structures. Cytogenetic markers anchored in these regions constitute a platform for heterologous mapping of legume genomes.
窄叶羽扇豆()最近被视为豆科参考物种。已经开发了遗传资源,包括基因组序列草图、连锁图谱、核DNA文库和细胞遗传学染色体特异性标记。在这里,我们采用了一种复杂的方法,包括DNA指纹图谱、测序、遗传图谱绘制和分子细胞遗传学,来定位和分析富含基因的区域(GRRs)。用基于短序列重复(SSR)的探针筛选了一个基因组细菌人工染色体(BAC)文库。对选定的BAC进行指纹图谱分析并组装成重叠群。BAC末端序列(BES)注释使我们能够选择用于测序的克隆,以GRRs为目标。此外,将BES与基因组序列的支架进行比对。遗传图谱补充了35个源自BES的标记,分布在14个连锁群中,并标记了37个支架。鉴定出的GRRs平均基因密度为19.6个基因/100 kb,物理与遗传距离比为11至109 kb/cM。多BAC荧光原位杂交(FISH)支持物理和遗传图谱绘制,并将五个新的连锁群定位到染色体上。确定了与五个豆科物种(,,,和)的基因组序列的共线性联系。对两个最大的羽扇豆GRRs的比较图谱分析为所有研究物种中的古代重复提供了新证据。这些区域在蝶形花亚科主要分支的代表中是保守的。此外,尽管豆科植物进化复杂,但核基因组的一些片段没有发生实质性改变,并保留了它们的准祖先结构。锚定在这些区域的细胞遗传学标记构成了豆科基因组异源图谱绘制的平台。