Su Zhenqiang, Fang Hong, Hong Huixiao, Shi Leming, Zhang Wenqian, Zhang Wenwei, Zhang Yanyan, Dong Zirui, Lancashire Lee J, Bessarabova Marina, Yang Xi, Ning Baitang, Gong Binsheng, Meehan Joe, Xu Joshua, Ge Weigong, Perkins Roger, Fischer Matthias, Tong Weida
Genome Biol. 2014 Dec 3;15(12):523. doi: 10.1186/s13059-014-0523-y.
Gene expression microarray has been the primary biomarker platform ubiquitously applied in biomedical research, resulting in enormous data, predictive models, and biomarkers accrued. Recently, RNA-seq has looked likely to replace microarrays, but there will be a period where both technologies co-exist. This raises two important questions: Can microarray-based models and biomarkers be directly applied to RNA-seq data? Can future RNA-seq-based predictive models and biomarkers be applied to microarray data to leverage past investment?
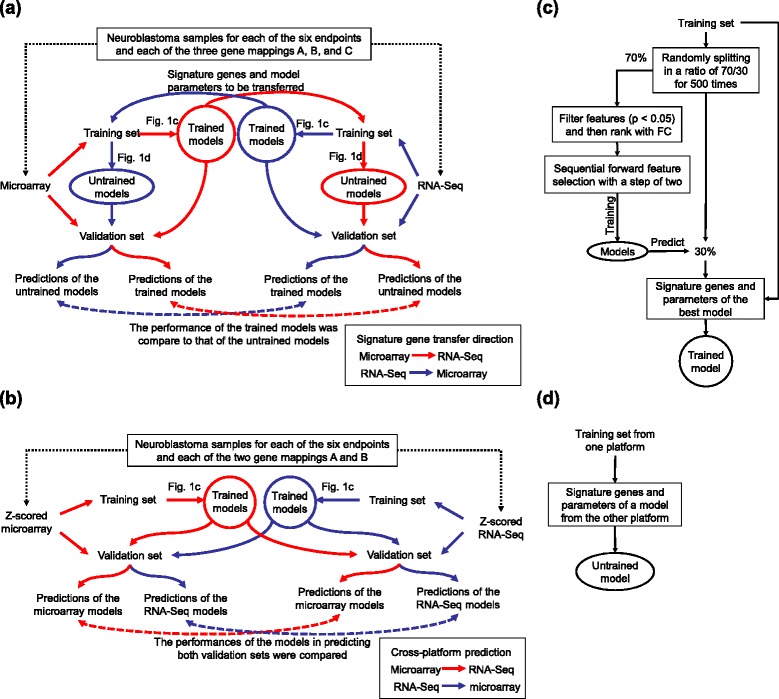
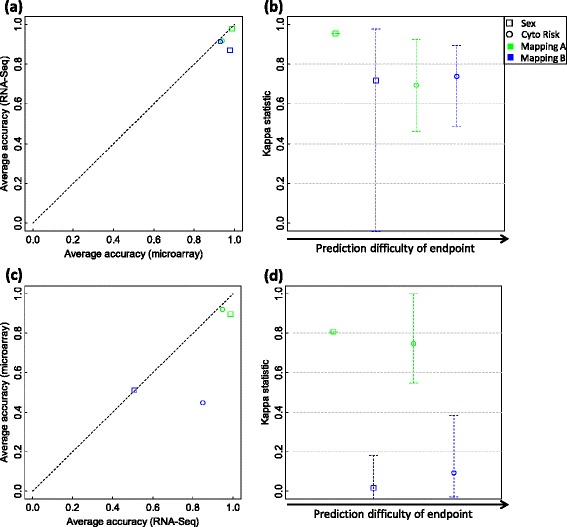
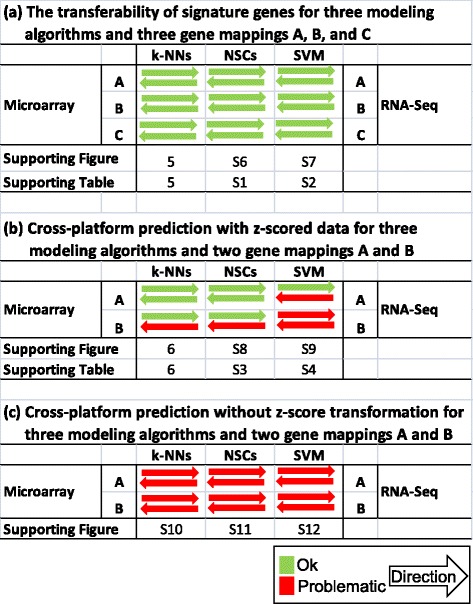
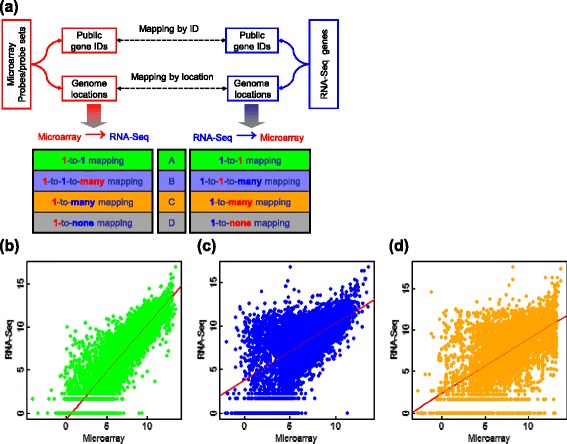
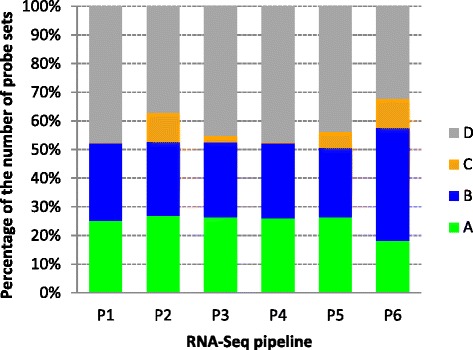
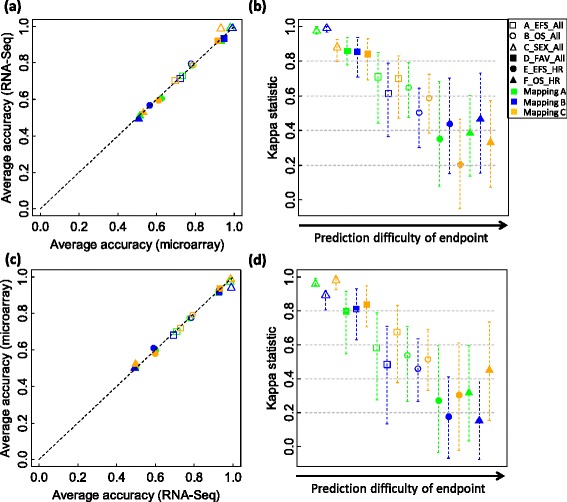
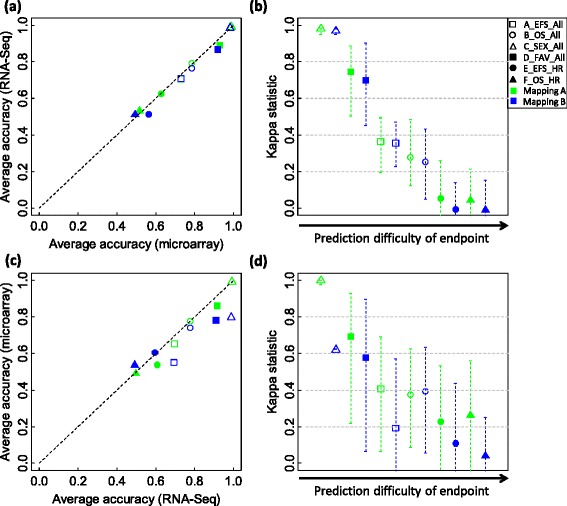
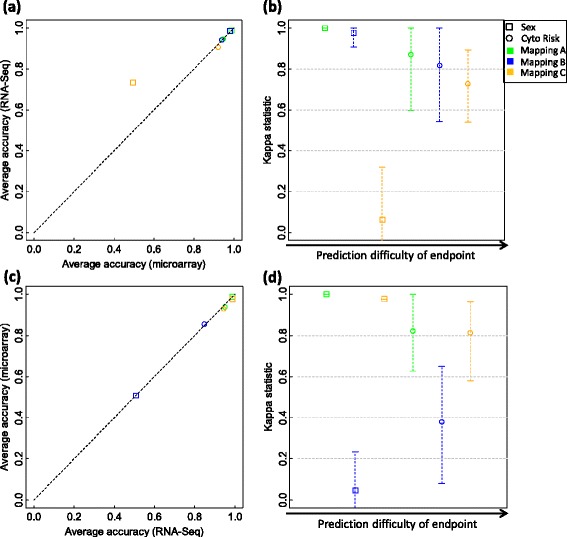
We systematically evaluated the transferability of predictive models and signature genes between microarray and RNA-seq using two large clinical data sets. The complexity of cross-platform sequence correspondence was considered in the analysis and examined using three human and two rat data sets, and three levels of mapping complexity were revealed. Three algorithms representing different modeling complexity were applied to the three levels of mappings for each of the eight binary endpoints and Cox regression was used to model survival times with expression data. In total, 240,096 predictive models were examined.
Signature genes of predictive models are reciprocally transferable between microarray and RNA-seq data for model development, and microarray-based models can accurately predict RNA-seq-profiled samples; while RNA-seq-based models are less accurate in predicting microarray-profiled samples and are affected both by the choice of modeling algorithm and the gene mapping complexity. The results suggest continued usefulness of legacy microarray data and established microarray biomarkers and predictive models in the forthcoming RNA-seq era.
基因表达微阵列一直是生物医学研究中广泛应用的主要生物标志物平台,产生了大量的数据、预测模型和积累的生物标志物。最近,RNA测序似乎有可能取代微阵列,但在一段时间内这两种技术将共存。这就引出了两个重要问题:基于微阵列的模型和生物标志物能否直接应用于RNA测序数据?未来基于RNA测序的预测模型和生物标志物能否应用于微阵列数据以利用过去的投资?
我们使用两个大型临床数据集系统地评估了微阵列和RNA测序之间预测模型和特征基因的可转移性。在分析中考虑了跨平台序列对应关系的复杂性,并使用三个人类和两个大鼠数据集进行了检验,揭示了三个映射复杂性水平。将代表不同建模复杂性的三种算法应用于八个二元终点中每个终点的三个映射水平,并使用Cox回归对表达数据的生存时间进行建模。总共检查了240,096个预测模型。
预测模型的特征基因在微阵列和RNA测序数据之间可相互转移用于模型开发,基于微阵列的模型可以准确预测RNA测序分析的样本;而基于RNA测序的模型在预测微阵列分析的样本时准确性较低,并且受到建模算法选择和基因映射复杂性的影响。结果表明,在即将到来的RNA测序时代,传统微阵列数据以及已建立的微阵列生物标志物和预测模型仍将有用。