Chekouo Thierry, Stingo Francesco C, Doecke James D, Do Kim-Anh
Department of Biostatistics, The University of Texas MD Anderson Cancer Center, Houston, 1400 Pressler Street, Unit 1411, Texas, 77030-3722, U.S.A.
CSIRO Computational Informatics/Australian e-Health Research Centre Level 5, UQ Health Sciences Building, 901/16 Royal Brisbane, Queensland, 4029, Australia.
Biometrics. 2015 Jun;71(2):428-38. doi: 10.1111/biom.12266. Epub 2015 Jan 30.
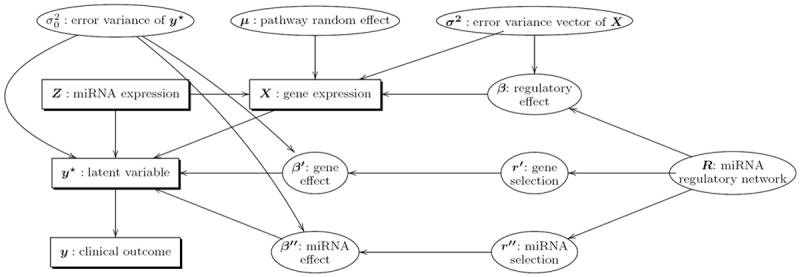
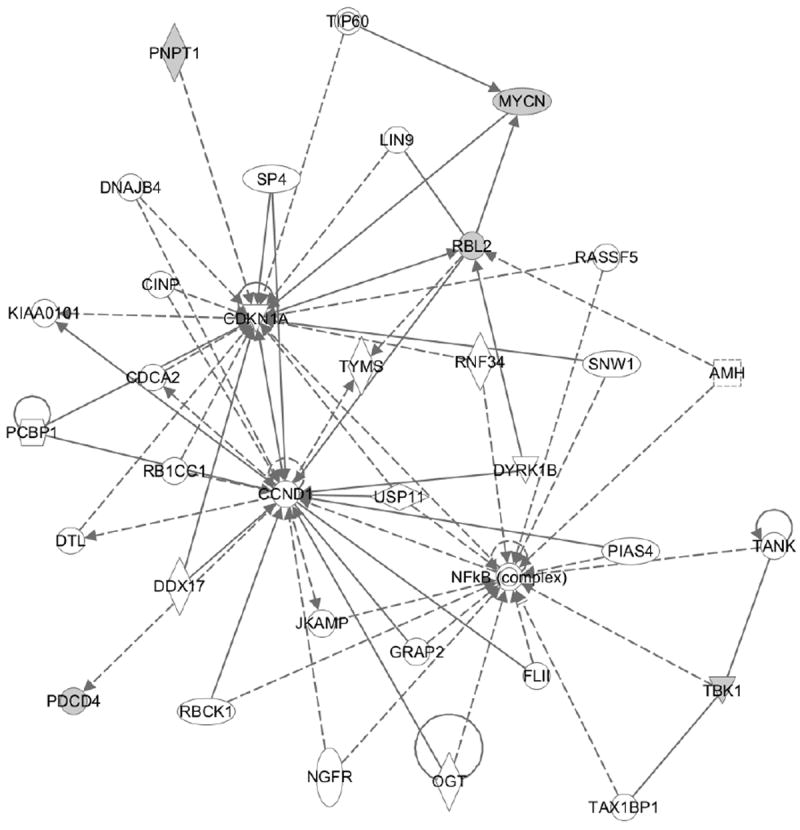
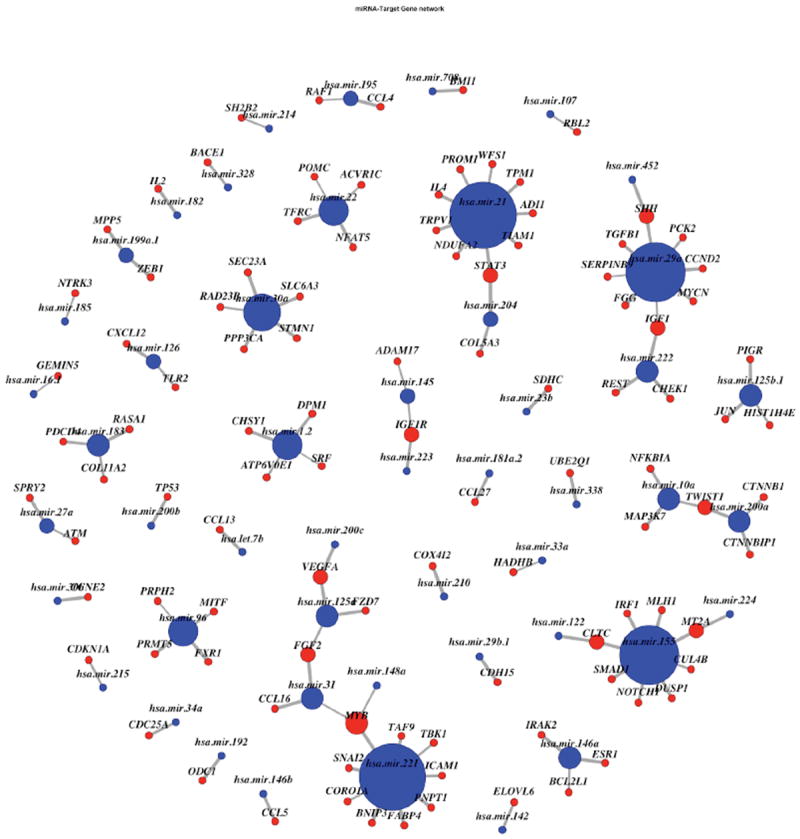
The availability of cross-platform, large-scale genomic data has enabled the investigation of complex biological relationships for many cancers. Identification of reliable cancer-related biomarkers requires the characterization of multiple interactions across complex genetic networks. MicroRNAs are small non-coding RNAs that regulate gene expression; however, the direct relationship between a microRNA and its target gene is difficult to measure. We propose a novel Bayesian model to identify microRNAs and their target genes that are associated with survival time by incorporating the microRNA regulatory network through prior distributions. We assume that biomarkers involved in regulatory networks are likely associated with survival time. We employ non-local prior distributions and a stochastic search method for the selection of biomarkers associated with the survival outcome. We use KEGG pathway information to incorporate correlated gene effects within regulatory networks. Using simulation studies, we assess the performance of our method, and apply it to experimental data of kidney renal cell carcinoma (KIRC) obtained from The Cancer Genome Atlas. Our novel method validates previously identified cancer biomarkers and identifies biomarkers specific to KIRC progression that were not previously discovered. Using the KIRC data, we confirm that biomarkers involved in regulatory networks are more likely to be associated with survival time, showing connections in one regulatory network for five out of six such genes we identified.
跨平台大规模基因组数据的可用性使得对多种癌症复杂生物学关系的研究成为可能。识别可靠的癌症相关生物标志物需要对复杂遗传网络中的多种相互作用进行表征。微小RNA是调节基因表达的小非编码RNA;然而,微小RNA与其靶基因之间的直接关系很难测量。我们提出了一种新颖的贝叶斯模型,通过先验分布纳入微小RNA调控网络,来识别与生存时间相关的微小RNA及其靶基因。我们假设参与调控网络的生物标志物可能与生存时间相关。我们采用非局部先验分布和随机搜索方法来选择与生存结果相关的生物标志物。我们利用KEGG通路信息来纳入调控网络内的相关基因效应。通过模拟研究,我们评估了我们方法的性能,并将其应用于从癌症基因组图谱获得的肾透明细胞癌(KIRC)实验数据。我们的新方法验证了先前鉴定的癌症生物标志物,并鉴定出了先前未发现的KIRC进展特异性生物标志物。利用KIRC数据,我们证实参与调控网络的生物标志物更有可能与生存时间相关,在我们鉴定的六个此类基因中的五个基因中,显示出在一个调控网络中的联系。