Škunca Nives, Dessimoz Christophe
ETH Zürich, Department of Computer Science, Universitätstr. 19, 8092 Zürich, Switzerland; Swiss Institute of Bioinformatics, Universitätstr. 6, 8092 Zürich, Switzerland; University College London, Gower St, London WC1E 6BT, UK.
University College London, Gower St, London WC1E 6BT, UK; Swiss Institute of Bioinformatics, Universitätstr. 6, 8092 Zürich, Switzerland.
PLoS One. 2015 Feb 13;10(2):e0114701. doi: 10.1371/journal.pone.0114701. eCollection 2015.
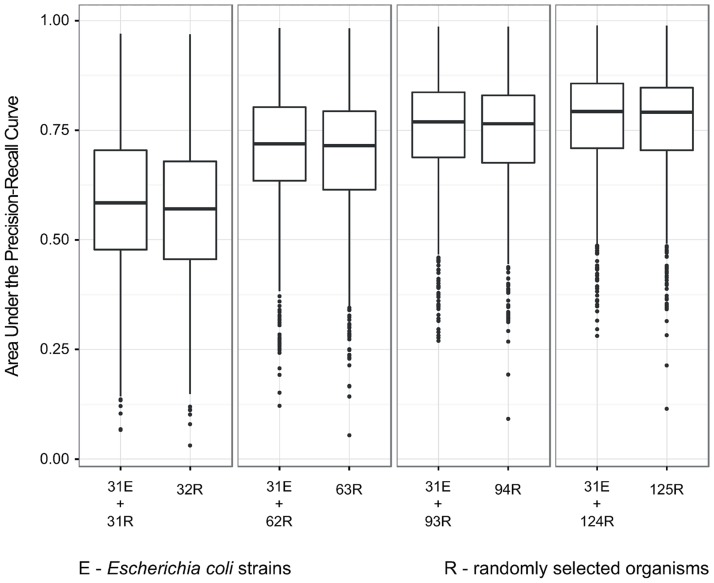
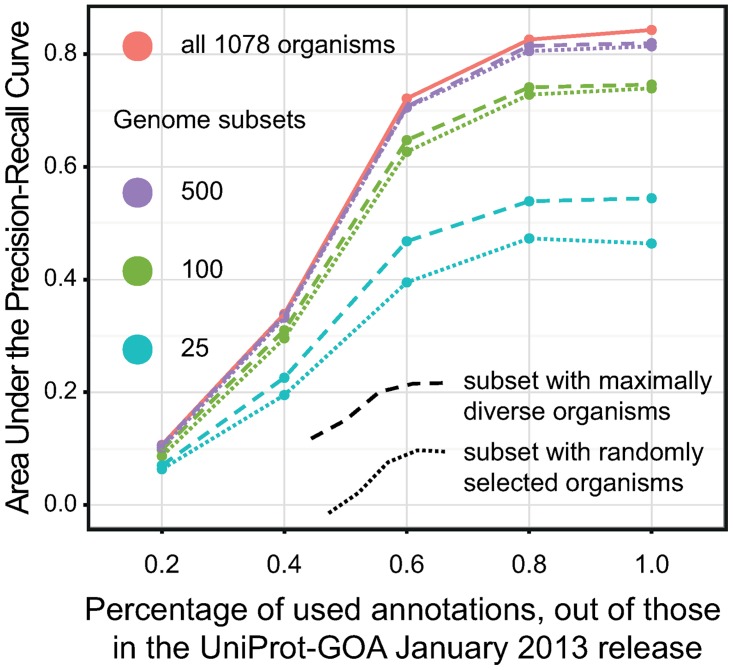
Phylogenetic profiling is a well-established approach for predicting gene function based on patterns of gene presence and absence across species. Much of the recent developments have focused on methodological improvements, but relatively little is known about the effect of input data size on the quality of predictions. In this work, we ask: how many genomes and functional annotations need to be considered for phylogenetic profiling to be effective? Phylogenetic profiling generally benefits from an increased amount of input data. However, by decomposing this improvement in predictive accuracy in terms of the contribution of additional genomes and of additional annotations, we observed diminishing returns in adding more than ∼ 100 genomes, whereas increasing the number of annotations remained strongly beneficial throughout. We also observed that maximising phylogenetic diversity within a clade of interest improves predictive accuracy, but the effect is small compared to changes in the number of genomes under comparison. Finally, we show that these findings are supported in light of the Open World Assumption, which posits that functional annotation databases are inherently incomplete. All the tools and data used in this work are available for reuse from http://lab.dessimoz.org/14_phylprof. Scripts used to analyse the data are available on request from the authors.
系统发育谱分析是一种基于物种间基因存在与否模式来预测基因功能的成熟方法。近期的许多进展都集中在方法改进上,但对于输入数据大小对预测质量的影响却知之甚少。在这项工作中,我们提出问题:为了使系统发育谱分析有效,需要考虑多少个基因组和功能注释?系统发育谱分析通常受益于输入数据量的增加。然而,通过从额外基因组和额外注释的贡献角度分解预测准确性的这种提高,我们观察到,增加超过约100个基因组时收益递减,而增加注释数量在整个过程中仍然非常有益。我们还观察到,在感兴趣的进化枝内最大化系统发育多样性可提高预测准确性,但与所比较基因组数量的变化相比,这种影响较小。最后,我们表明,根据开放世界假设,这些发现得到了支持,该假设认为功能注释数据库本质上是不完整的。这项工作中使用的所有工具和数据都可从http://lab.dessimoz.org/14_phylprof重复使用。用于分析数据的脚本可应作者要求提供。