Mensaert Klaas, Van Criekinge Wim, Thas Olivier, Schuuring Ed, Steenbergen Renske D M, Wisman G Bea A, De Meyer Tim
Department of Mathematical Modeling, Statistics and Bioinformatics, Ghent University Ghent, Belgium.
Department of Mathematical Modeling, Statistics and Bioinformatics, Ghent University Ghent, Belgium ; Department of Pathology, VU University Medical Center Amsterdam, Netherlands.
Front Genet. 2015 Feb 4;6:16. doi: 10.3389/fgene.2015.00016. eCollection 2015.
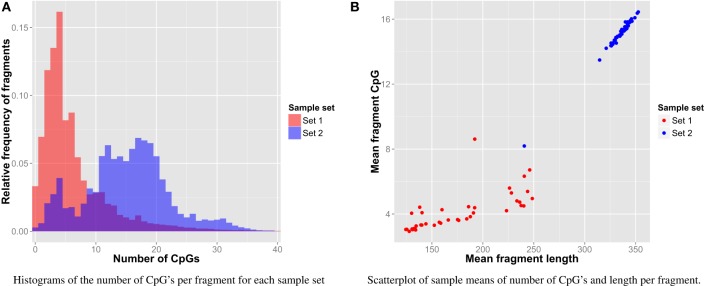
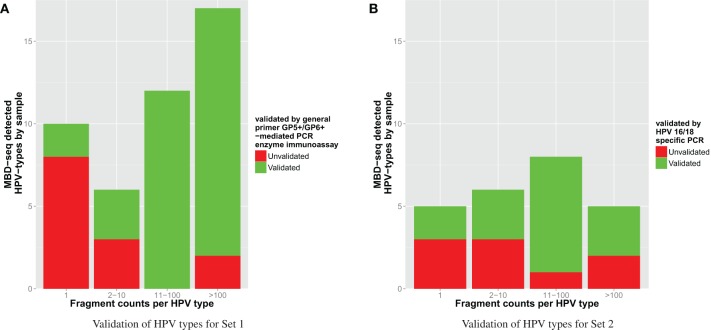
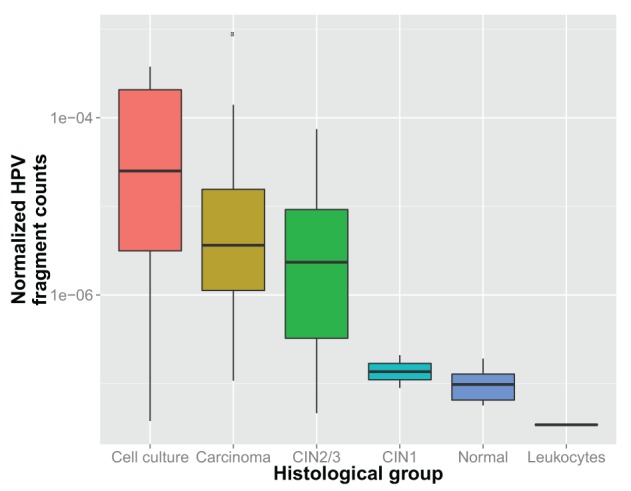
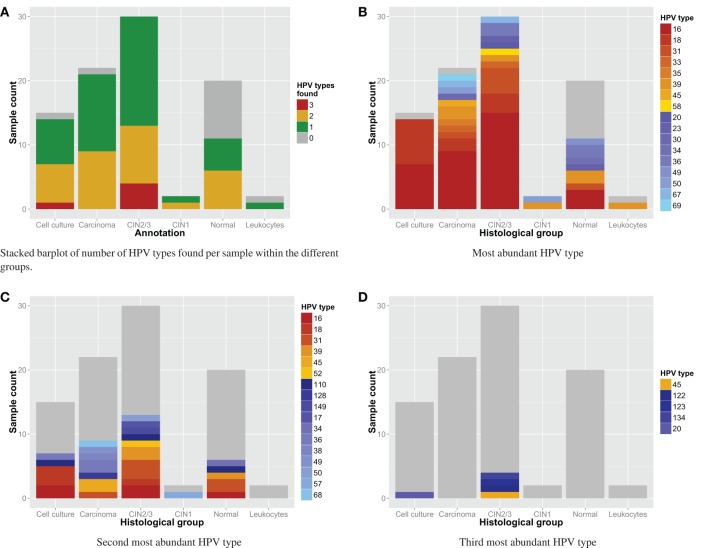
Most next generation sequencing experiments generate more data than is usable for the experimental set up. For example, methyl-CpG binding domain (MBD) affinity purification based sequencing is often used for DNA-methylation profiling, but up to 30% of the sequenced fragments cannot be mapped uniquely to the reference genome. Here we present and evaluate a methodology for the identification of viruses in these otherwise unused paired-end MBD-seq data. Viral detection is accomplished by mapping non-reference alignable reads to a comprehensive set of viral genomes. As viruses play an important role in epigenetics and cancer development, 92 (pre)malignant and benign samples, originating from two different collections of cervical samples and related cell lines, were used in this study. These samples include primary carcinomas (n = 22), low- and high-grade cervical intraepithelial neoplasia (CIN1 and CIN2/3 - n = 2/n = 30) and normal tissue (n = 20), as well as control samples (n = 17). Viruses that were detected include phages, adenoviruses, herpesviridae and HPV. HPV, which causes virtually all cervical cancers, was identified in 95% of the carcinomas, 100% of the CIN2/3 samples, both CIN1 samples and in 55% of the normal samples. Comparing the amount of mapped fragments on HPV for each HPV-infected sample yielded a significant difference between normal samples and carcinomas or CIN2/3 samples (adjusted p-values resp. <10(-5), <10(-5)), reflecting different viral loads and/or methylation degrees in non-normal samples. Fragments originating from different HPV types could be distinguished and were independently validated by PCR-based assays in 71% of the detections. In conclusion, although limited by the a priori knowledge of viral reference genome sequences, the proposed methodology can provide a first confined but substantial insight into the presence, concentration and types of methylated viral sequences in MBD-seq data at low additional cost.
大多数下一代测序实验产生的数据量超过了实验设置可使用的范围。例如,基于甲基化CpG结合域(MBD)亲和纯化的测序常用于DNA甲基化分析,但高达30%的测序片段无法唯一地映射到参考基因组。在此,我们提出并评估一种从这些原本未使用的双末端MBD-seq数据中鉴定病毒的方法。病毒检测是通过将非参考可比对的 reads 映射到一套全面的病毒基因组来完成的。由于病毒在表观遗传学和癌症发展中起着重要作用,本研究使用了来自两个不同宫颈样本及相关细胞系集合的92个(癌前)恶性和良性样本。这些样本包括原发性癌(n = 22)、低级别和高级别宫颈上皮内瘤变(CIN1和CIN2/3 - n = 2/n = 30)以及正常组织(n = 20),还有对照样本(n = 17)。检测到的病毒包括噬菌体、腺病毒、疱疹病毒科和人乳头瘤病毒(HPV)。几乎所有宫颈癌都是由HPV引起的,在95%的癌组织、100%的CIN2/3样本、两个CIN1样本以及55%的正常样本中都检测到了HPV。比较每个HPV感染样本上HPV的映射片段数量,发现正常样本与癌组织或CIN2/3样本之间存在显著差异(调整后的p值分别<10^(-5),<10^(-5)),这反映了非正常样本中不同的病毒载量和/或甲基化程度。在71%的检测中,可以区分来自不同HPV类型的片段,并通过基于PCR的检测进行独立验证。总之,尽管该方法受限于病毒参考基因组序列的先验知识,但以较低的额外成本,能够对MBD-seq数据中甲基化病毒序列的存在、浓度和类型提供初步但有限且重要的见解。