Tasnim Masruba, Ma Shining, Yang Ei-Wen, Jiang Tao, Li Wei
BMC Genomics. 2015;16 Suppl 2(Suppl 2):S15. doi: 10.1186/1471-2164-16-S2-S15. Epub 2015 Jan 21.
RNA-Seq based transcriptome assembly has become a fundamental technique for studying expressed mRNAs (i.e., transcripts or isoforms) in a cell using high-throughput sequencing technologies, and is serving as a basis to analyze the structural and quantitative differences of expressed isoforms between samples. However, the current transcriptome assembly algorithms are not specifically designed to handle large amounts of errors that are inherent in real RNA-Seq datasets, especially those involving multiple samples, making downstream differential analysis applications difficult. On the other hand, multiple sample RNA-Seq datasets may provide more information than single sample datasets that can be utilized to improve the performance of transcriptome assembly and abundance estimation, but such information remains overlooked by the existing assembly tools.
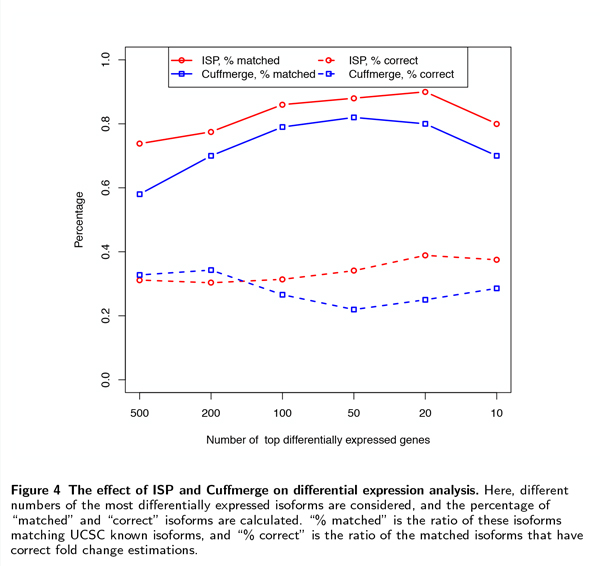
We formulate a computational framework of transcriptome assembly that is capable of handling noisy RNA-Seq reads and multiple sample RNA-Seq datasets efficiently. We show that finding an optimal solution under this framework is an NP-hard problem. Instead, we develop an efficient heuristic algorithm, called Iterative Shortest Path (ISP), based on linear programming (LP) and integer linear programming (ILP). Our preliminary experimental results on both simulated and real datasets and comparison with the existing assembly tools demonstrate that (i) the ISP algorithm is able to assemble transcriptomes with a greatly increased precision while keeping the same level of sensitivity, especially when many samples are involved, and (ii) its assembly results help improve downstream differential analysis. The source code of ISP is freely available at http://alumni.cs.ucr.edu/~liw/isp.html.
基于RNA测序的转录组组装已成为利用高通量测序技术研究细胞中表达的mRNA(即转录本或异构体)的一项基础技术,并为分析样本间表达异构体的结构和数量差异提供了依据。然而,当前的转录组组装算法并非专门设计用于处理真实RNA测序数据集中固有的大量错误,尤其是那些涉及多个样本的数据集,这使得下游差异分析应用变得困难。另一方面,多样本RNA测序数据集可能比单样本数据集提供更多信息,可用于提高转录组组装和丰度估计的性能,但现有组装工具却忽略了这些信息。
我们制定了一个转录组组装的计算框架,该框架能够有效处理有噪声的RNA测序读段和多样本RNA测序数据集。我们表明,在此框架下找到最优解是一个NP难问题。相反,我们基于线性规划(LP)和整数线性规划(ILP)开发了一种高效的启发式算法,称为迭代最短路径(ISP)。我们在模拟数据集和真实数据集上的初步实验结果以及与现有组装工具的比较表明:(i)ISP算法能够在保持相同灵敏度水平的同时,以显著提高的精度组装转录组,尤其是在涉及多个样本时;(ii)其组装结果有助于改进下游差异分析。ISP 的源代码可在http://alumni.cs.ucr.edu/~liw/isp.html免费获取。