Lim Junghyun, Lachenmayer M Lenard, Wu Shuai, Liu Wenchao, Kundu Mondira, Wang Rong, Komatsu Masaaki, Oh Young J, Zhao Yanxiang, Yue Zhenyu
Department of Neurology and Neuroscience, Friedman Brain Institute, Icahn School of Medicine at Mount Sinai, New York, New York, United States of America.
Department of Applied Biology and Chemical Technology, State Key Laboratory of Chirosciences, The Hong Kong Polytechnic University, Hung Hom, Kowloon, Hong Kong, China.
PLoS Genet. 2015 Feb 27;11(2):e1004987. doi: 10.1371/journal.pgen.1004987. eCollection 2015.
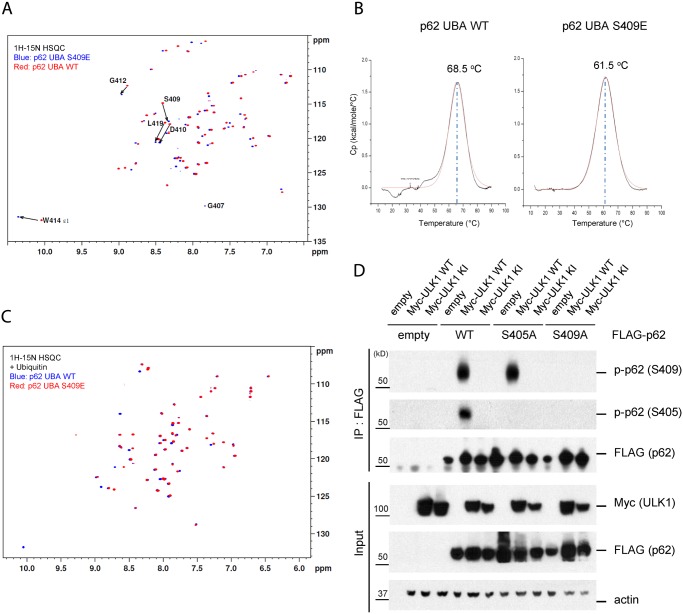
Disruption of proteostasis, or protein homeostasis, is often associated with aberrant accumulation of misfolded proteins or protein aggregates. Autophagy offers protection to cells by removing toxic protein aggregates and injured organelles in response to proteotoxic stress. However, the exact mechanism whereby autophagy recognizes and degrades misfolded or aggregated proteins has yet to be elucidated. Mounting evidence demonstrates the selectivity of autophagy, which is mediated through autophagy receptor proteins (e.g. p62/SQSTM1) linking autophagy cargos and autophagosomes. Here we report that proteotoxic stress imposed by the proteasome inhibition or expression of polyglutamine expanded huntingtin (polyQ-Htt) induces p62 phosphorylation at its ubiquitin-association (UBA) domain that regulates its binding to ubiquitinated proteins. We find that autophagy-related kinase ULK1 phosphorylates p62 at a novel phosphorylation site S409 in UBA domain. Interestingly, phosphorylation of p62 by ULK1 does not occur upon nutrient starvation, in spite of its role in canonical autophagy signaling. ULK1 also phosphorylates S405, while S409 phosphorylation critically regulates S405 phosphorylation. We find that S409 phosphorylation destabilizes the UBA dimer interface, and increases binding affinity of p62 to ubiquitin. Furthermore, lack of S409 phosphorylation causes accumulation of p62, aberrant localization of autophagy proteins and inhibition of the clearance of ubiquitinated proteins or polyQ-Htt. Therefore, our data provide mechanistic insights into the regulation of selective autophagy by ULK1 and p62 upon proteotoxic stress. Our study suggests a potential novel drug target in developing autophagy-based therapeutics for the treatment of proteinopathies including Huntington's disease.
蛋白质稳态(即蛋白质平衡)的破坏通常与错误折叠蛋白质或蛋白质聚集体的异常积累有关。自噬通过在蛋白毒性应激反应中清除有毒蛋白质聚集体和受损细胞器来为细胞提供保护。然而,自噬识别并降解错误折叠或聚集蛋白质的确切机制尚未阐明。越来越多的证据表明自噬具有选择性,这是通过自噬受体蛋白(如p62/SQSTM1)介导的,这些蛋白将自噬货物与自噬体连接起来。在此,我们报告蛋白酶体抑制或聚谷氨酰胺扩展的亨廷顿蛋白(polyQ-Htt)表达所施加的蛋白毒性应激会诱导p62在其泛素结合(UBA)结构域发生磷酸化,该磷酸化调节其与泛素化蛋白质的结合。我们发现自噬相关激酶ULK1在UBA结构域的一个新的磷酸化位点S409使p62磷酸化。有趣的是,尽管ULK1在经典自噬信号传导中起作用,但在营养饥饿时它不会使p62磷酸化。ULK1还使S405磷酸化,而S409磷酸化关键调节S405磷酸化。我们发现S409磷酸化使UBA二聚体界面不稳定,并增加p62与泛素的结合亲和力。此外,缺乏S409磷酸化会导致p62积累、自噬蛋白异常定位以及泛素化蛋白质或polyQ-Htt清除的抑制。因此,我们的数据为蛋白毒性应激时ULK1和p62对选择性自噬的调节提供了机制性见解。我们的研究表明在开发基于自噬的治疗方法以治疗包括亨廷顿舞蹈病在内的蛋白质病方面有一个潜在的新药物靶点。