Selvin David, Renaud Jean-Marc
Department of Cellular and Molecular Medicine, University of Ottawa, Ottawa, Ontario, Canada.
Department of Cellular and Molecular Medicine, University of Ottawa, Ottawa, Ontario, Canada
Physiol Rep. 2015 Mar;3(3). doi: 10.14814/phy2.12303.
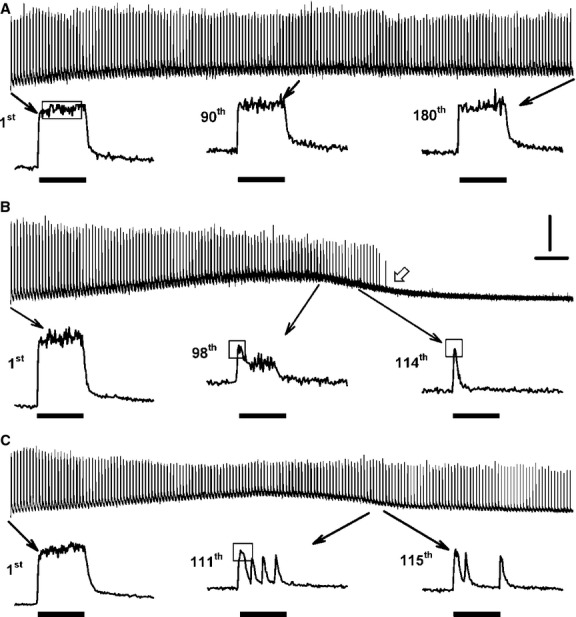
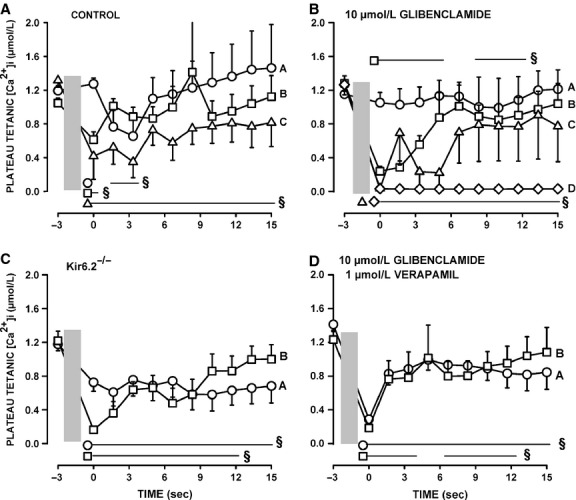
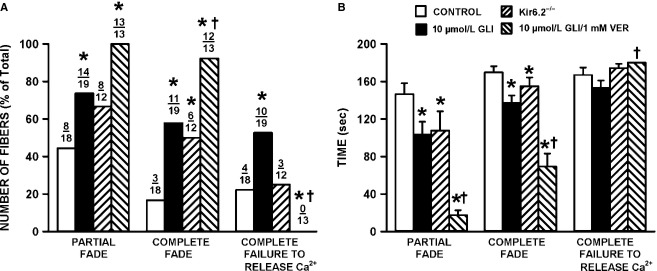
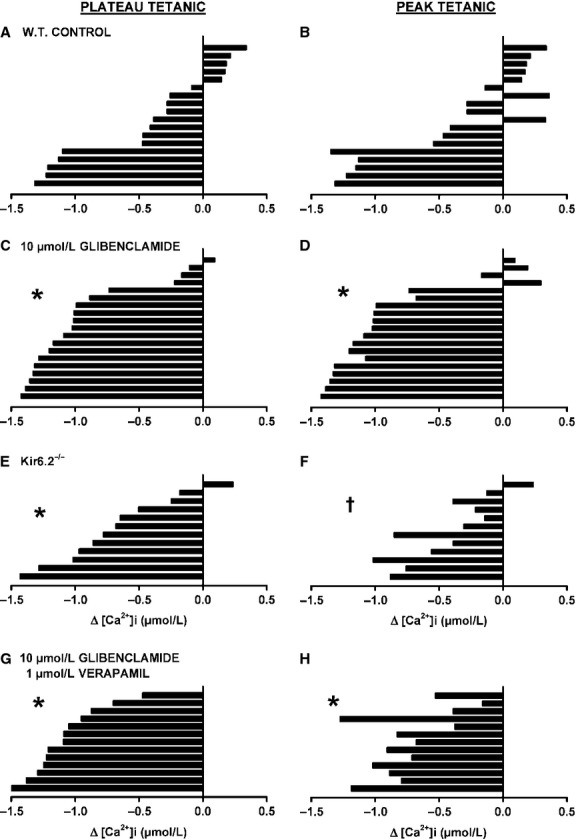
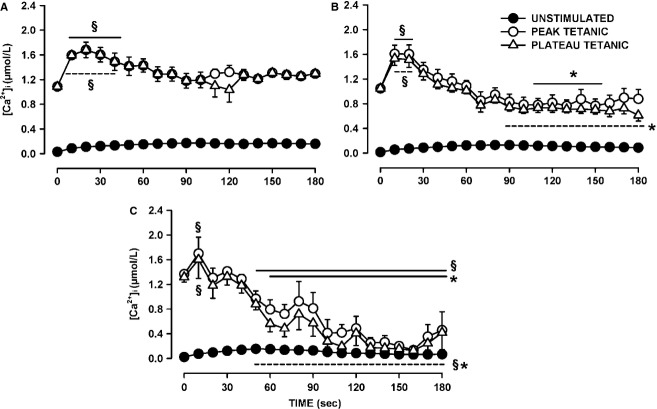
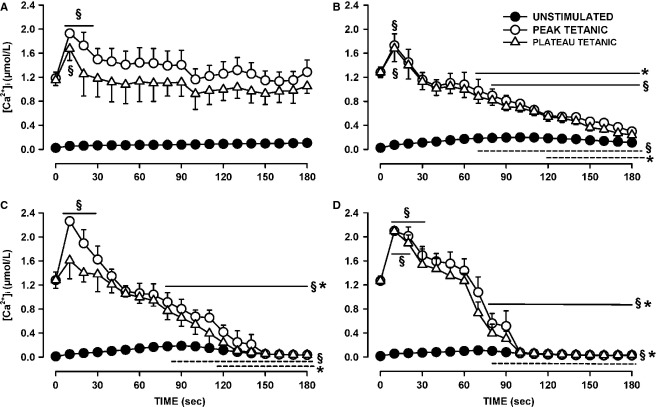
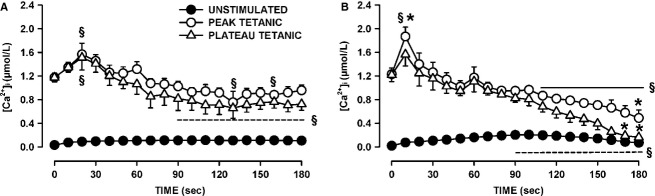
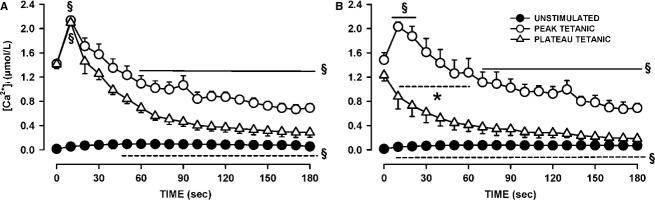
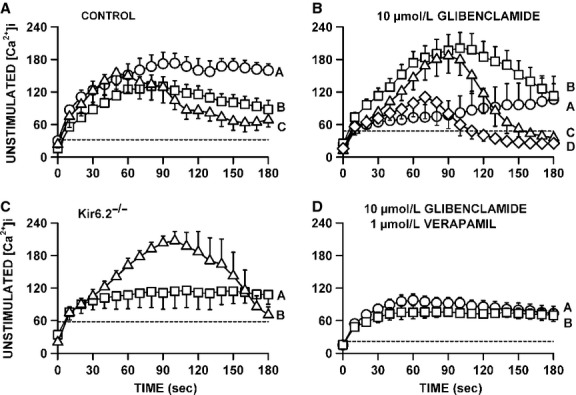
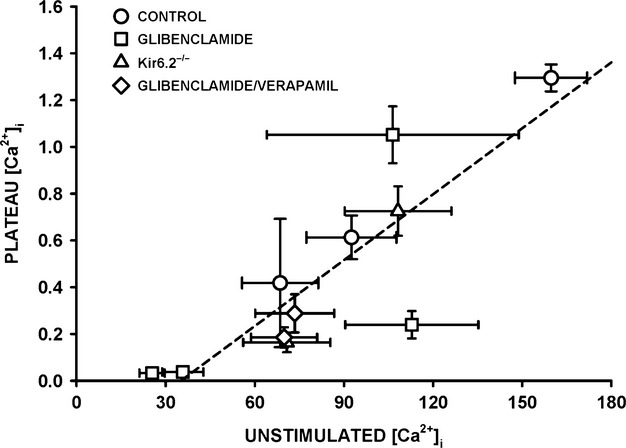
One objective of this study was to document how individual FDB muscle fibers depend on the myoprotection of KATP channels during fatigue. Verapamil, a CaV1.1 channel blocker, prevents large increases in unstimulated force during fatigue in KATP-channel-deficient muscles. A second objective was to determine if verapamil reduces unstimulated [Ca(2+)]i in KATP-channel-deficient fibers. We measured changes in myoplasmic [Ca(2+)] ([Ca(2+)]i) using two KATP-channel-deficient models: (1) a pharmacological approach exposing fibers to glibenclamide, a channel blocker, and (2) a genetic approach using fibers from null mice for the Kir6.2 gene. Fatigue was elicited with one tetanic contraction every sec for 3 min. For all conditions, large differences in fatigue kinetics were observed from fibers which had greater tetanic [Ca(2+)]i at the end than at the beginning of fatigue to fibers which eventually completely failed to release Ca(2+) upon stimulation. Compared to control conditions, KATP-channel-deficient fibers had a greater proportion of fiber with large decreases in tetanic [Ca(2+)]i, fade and complete failure to release Ca(2+) upon stimulation. There was, however, a group of KATP-channel-deficient fibers that had similar fatigue kinetics to those of the most fatigue-resistant control fibers. For the first time, differences in fatigue kinetics were observed between Kir6.2(-/-) and glibenclamide-exposed muscle fibers. Verapamil significantly reduced unstimulated and tetanic [Ca(2+)]i. It is concluded that not all fibers are dependent on the myoprotection of KATP channels and that the decrease in unstimulated force by verapamil reported in a previous studies in glibenclamide-exposed fibers is due to a reduction in Ca(2+) load by reducing Ca(2+) influx through CaV1.1 channels between and during contractions.
本研究的一个目的是记录在疲劳过程中,单个趾长伸肌(FDB)肌纤维如何依赖于ATP敏感性钾通道(KATP通道)的肌保护作用。维拉帕米是一种L型钙通道(CaV1.1通道)阻滞剂,可防止KATP通道缺陷型肌肉在疲劳过程中未受刺激时的力量大幅增加。第二个目的是确定维拉帕米是否能降低KATP通道缺陷型纤维中未受刺激时的胞内钙离子浓度([Ca(2+)]i)。我们使用两种KATP通道缺陷模型来测量肌浆钙离子浓度([Ca(2+)]i)的变化:(1)一种药理学方法,将纤维暴露于格列本脲(一种通道阻滞剂)中;(2)一种遗传学方法,使用来自Kir6.2基因敲除小鼠的纤维。通过每秒一次强直收缩,持续3分钟来引发疲劳。在所有条件下,从疲劳开始时强直收缩时[Ca(2+)]i较大到最终在刺激时完全无法释放Ca(2+)的纤维,观察到疲劳动力学存在很大差异。与对照条件相比,KATP通道缺陷型纤维中,强直收缩时[Ca(2+)]i大幅下降、出现疲劳以及在刺激时完全无法释放Ca(2+)的纤维比例更高。然而,有一组KATP通道缺陷型纤维,其疲劳动力学与最抗疲劳的对照纤维相似。首次观察到Kir6.2基因敲除(Kir6.2(-/-))的肌肉纤维和暴露于格列本脲的肌肉纤维在疲劳动力学上存在差异。维拉帕米显著降低了未受刺激时和强直收缩时的[Ca(2+)]i。得出的结论是,并非所有纤维都依赖于KATP通道的肌保护作用,并且先前在暴露于格列本脲的纤维中所报道的维拉帕米使未受刺激时的力量降低,是由于在收缩之间和收缩过程中通过减少CaV1.1通道的Ca(2+)内流,从而降低了Ca(2+)负荷。