Verschueren Erik, Von Dollen John, Cimermancic Peter, Gulbahce Natali, Sali Andrej, Krogan Nevan J
Department of Cellular & Molecular Pharmacology, University of California, San Francisco, San Francisco, California.
California Institute for Quantitative Biomedical Sciences, San Francisco, California.
Curr Protoc Bioinformatics. 2015 Mar 9;49:8.19.1-8.19.16. doi: 10.1002/0471250953.bi0819s49.
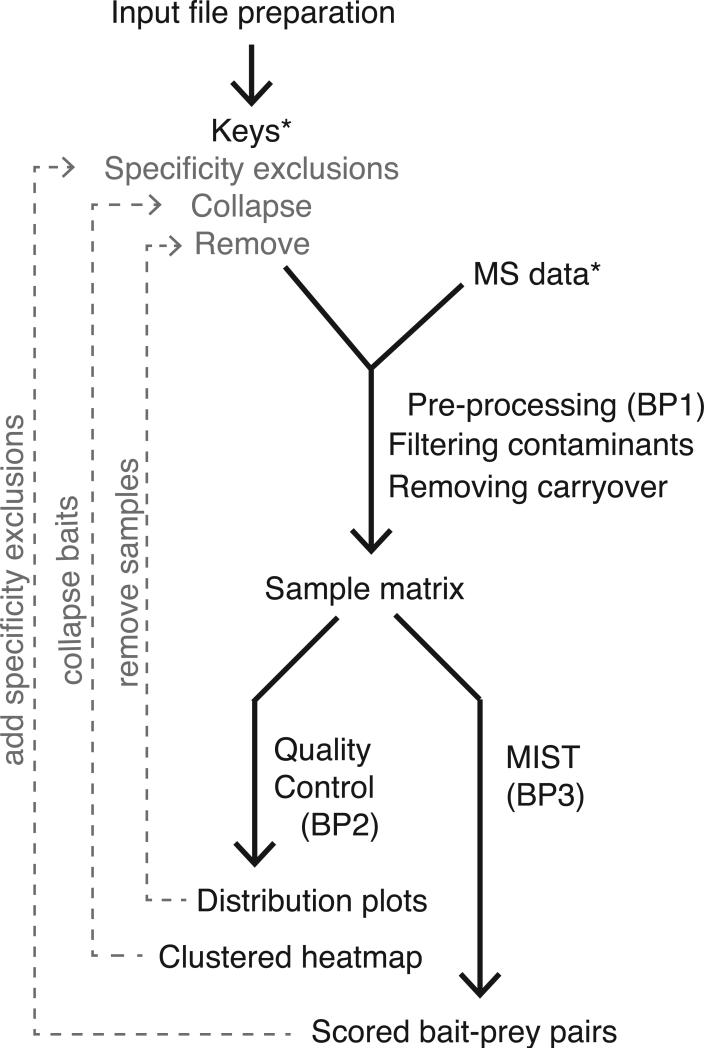
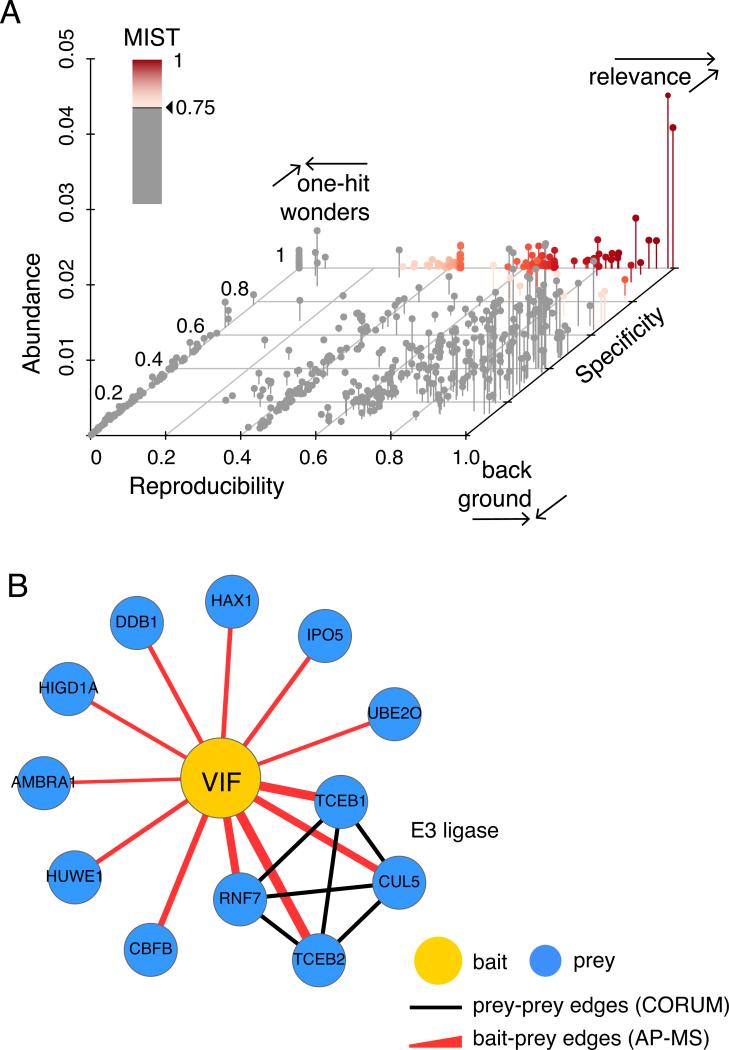
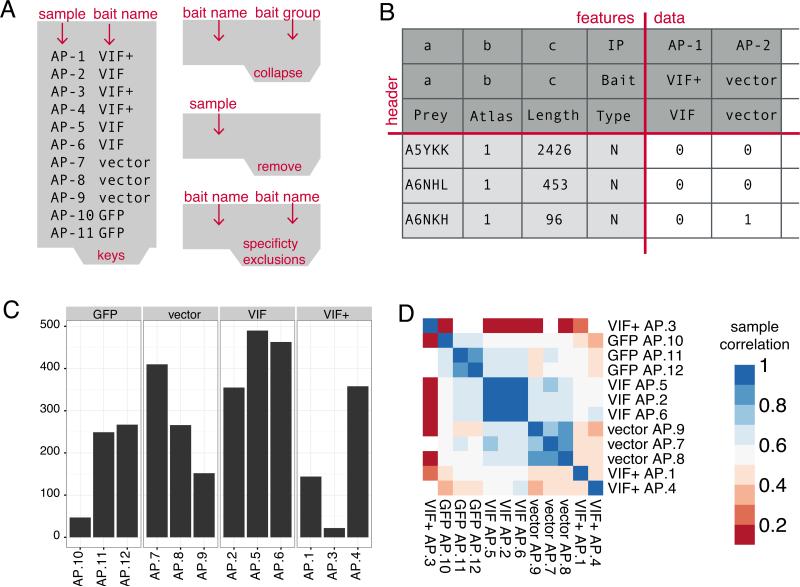
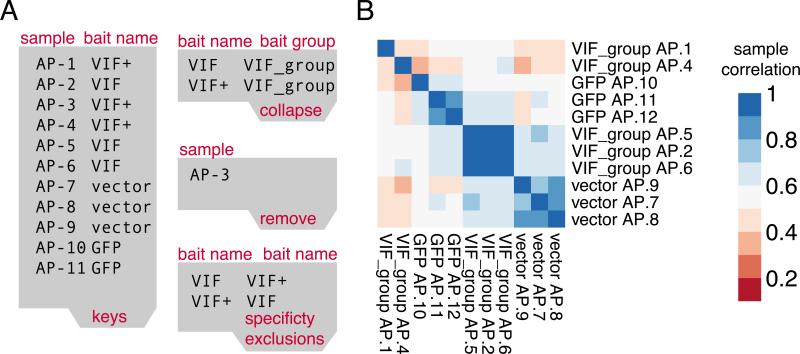
High-throughput Affinity Purification Mass Spectrometry (AP-MS) experiments can identify a large number of protein interactions, but only a fraction of these interactions are biologically relevant. Here, we describe a comprehensive computational strategy to process raw AP-MS data, perform quality controls, and prioritize biologically relevant bait-prey pairs in a set of replicated AP-MS experiments with Mass spectrometry interaction STatistics (MiST). The MiST score is a linear combination of prey quantity (abundance), abundance invariability across repeated experiments (reproducibility), and prey uniqueness relative to other baits (specificity). We describe how to run the full MiST analysis pipeline in an R environment and discuss a number of configurable options that allow the lay user to convert any large-scale AP-MS data into an interpretable, biologically relevant protein-protein interaction network.
高通量亲和纯化质谱(AP-MS)实验能够识别大量的蛋白质相互作用,但其中只有一小部分相互作用具有生物学相关性。在此,我们描述了一种全面的计算策略,用于处理原始AP-MS数据、进行质量控制,并在一组使用质谱相互作用统计(MiST)的重复AP-MS实验中,对具有生物学相关性的诱饵-猎物对进行优先级排序。MiST分数是猎物数量(丰度)、重复实验中丰度的不变性(可重复性)以及猎物相对于其他诱饵的独特性(特异性)的线性组合。我们描述了如何在R环境中运行完整的MiST分析流程,并讨论了一些可配置选项,这些选项使普通用户能够将任何大规模AP-MS数据转化为一个可解释的、具有生物学相关性的蛋白质-蛋白质相互作用网络。