Campbell Melody G, Veesler David, Cheng Anchi, Potter Clinton S, Carragher Bridget
National Resource for Automated Molecular Microscopy, The Scripps Research Institute, La Jolla, United States.
Elife. 2015 Mar 11;4:e06380. doi: 10.7554/eLife.06380.
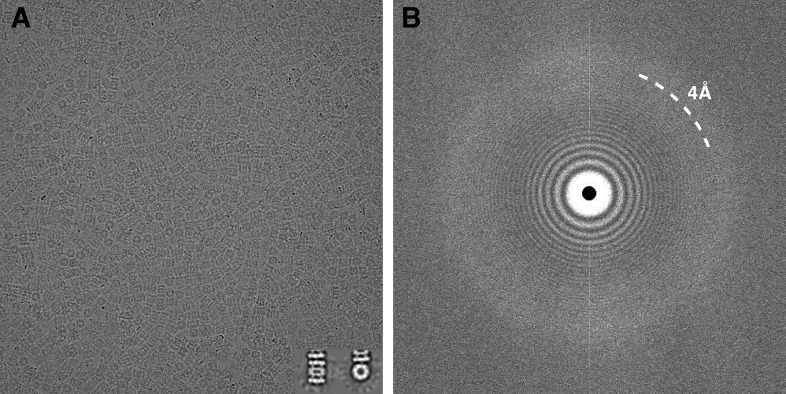
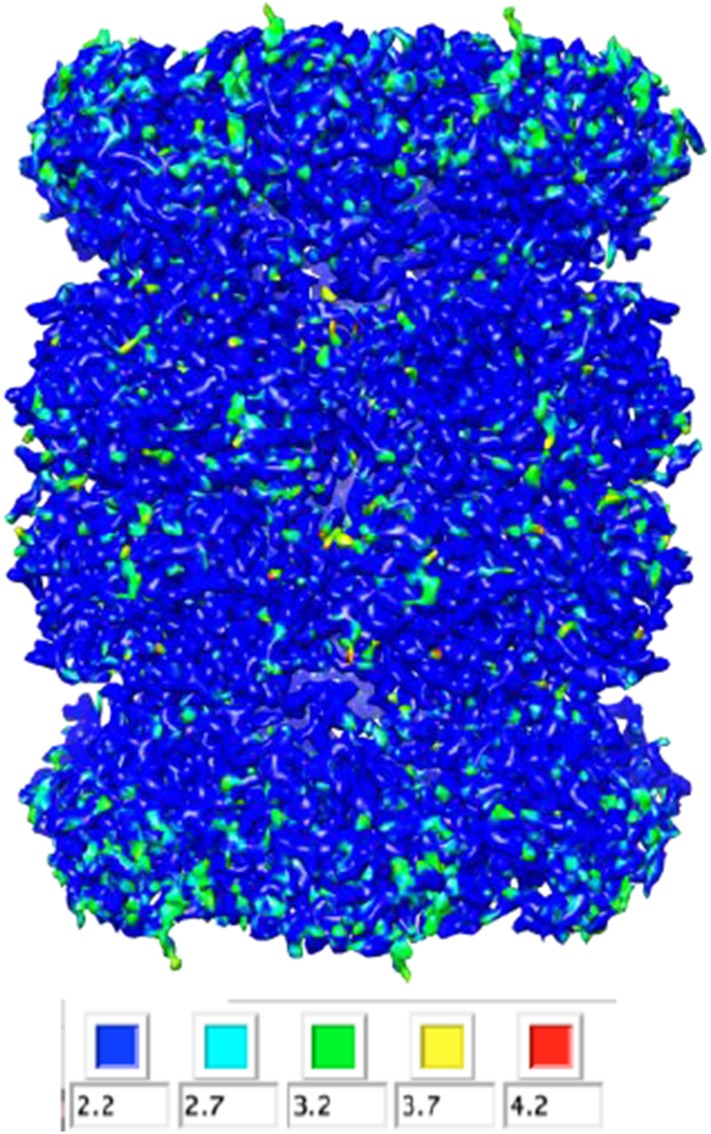
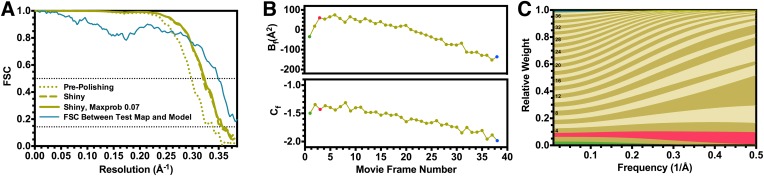
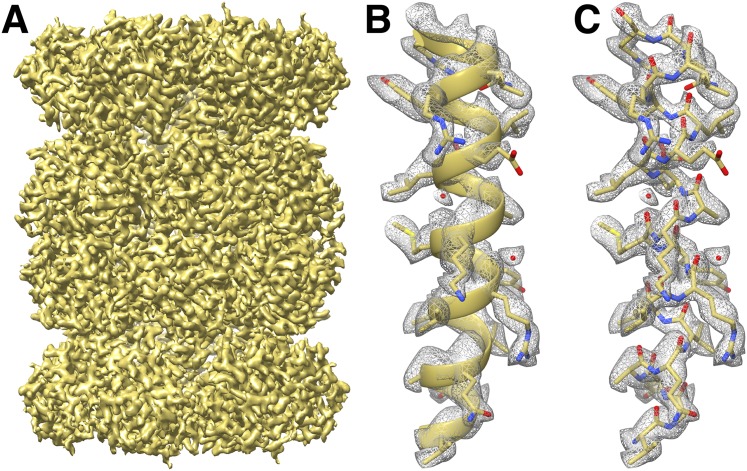
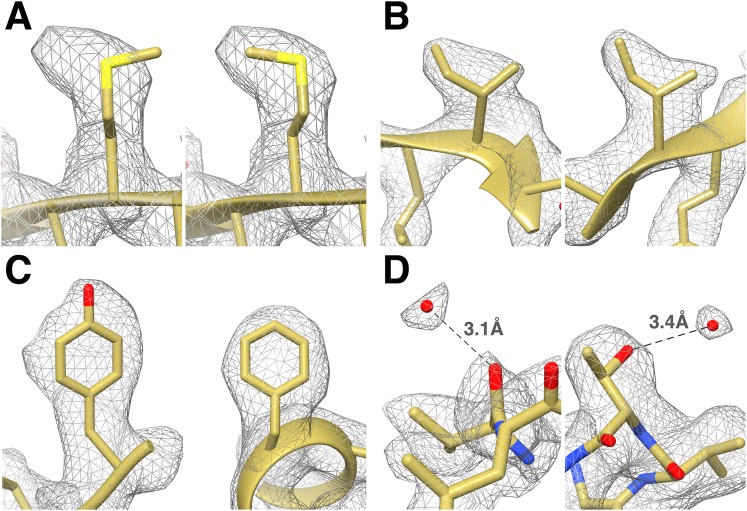
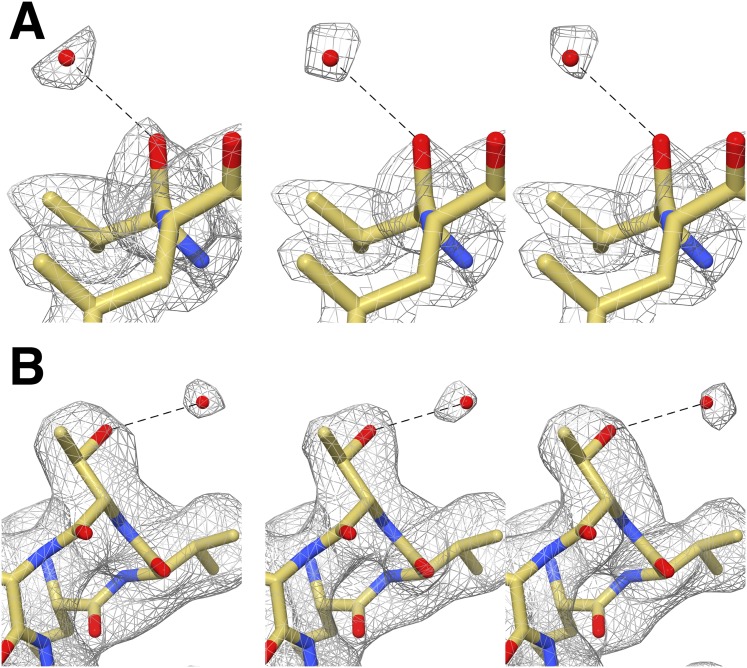
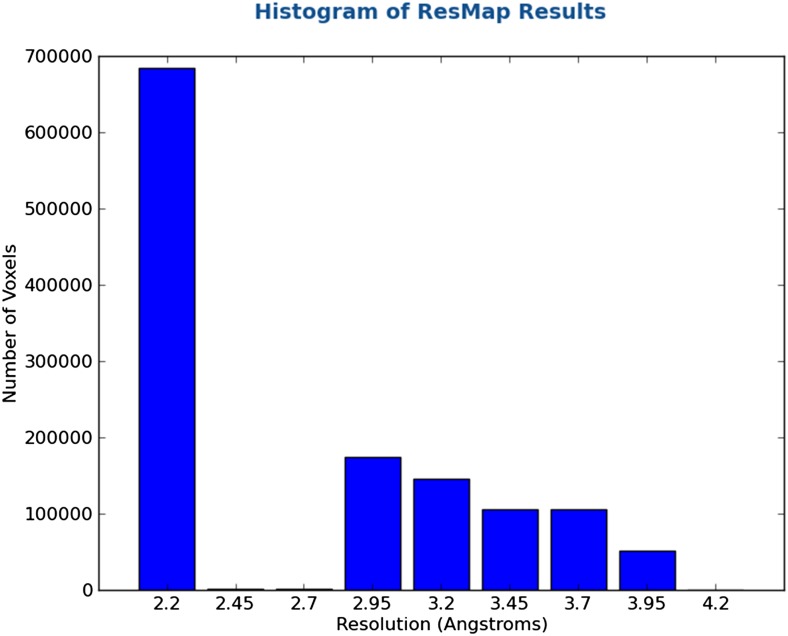
Recent developments in detector hardware and image-processing software have revolutionized single particle cryo-electron microscopy (cryoEM) and led to a wave of near-atomic resolution (typically ∼3.3 Å) reconstructions. Reaching resolutions higher than 3 Å is a prerequisite for structure-based drug design and for cryoEM to become widely interesting to pharmaceutical industries. We report here the structure of the 700 kDa Thermoplasma acidophilum 20S proteasome (T20S), determined at 2.8 Å resolution by single-particle cryoEM. The quality of the reconstruction enables identifying the rotameric conformation adopted by some amino-acid side chains (rotamers) and resolving ordered water molecules, in agreement with the expectations for crystal structures at similar resolutions. The results described in this manuscript demonstrate that single particle cryoEM is capable of competing with X-ray crystallography for determination of protein structures of suitable quality for rational drug design.
探测器硬件和图像处理软件的最新进展彻底改变了单颗粒冷冻电子显微镜技术(cryoEM),并引发了一波近原子分辨率(通常约为3.3 Å)的结构重建浪潮。达到高于3 Å的分辨率是基于结构的药物设计以及使冷冻电子显微镜技术受到制药行业广泛关注的先决条件。我们在此报告了嗜热栖热菌700 kDa 20S蛋白酶体(T20S)的结构,该结构通过单颗粒冷冻电子显微镜技术在2.8 Å分辨率下测定。重建质量能够识别一些氨基酸侧链所采用的旋转异构体构象(rotamers)并解析有序水分子,这与类似分辨率下晶体结构的预期相符。本手稿中描述的结果表明,单颗粒冷冻电子显微镜技术在确定适合合理药物设计的蛋白质结构方面能够与X射线晶体学相媲美。