The Keck Advanced Microscopy Laboratory, Department of Biochemistry and Biophysics, University of California, San Francisco (UCSF), San Francisco, California, USA.
Nat Methods. 2013 Jun;10(6):584-90. doi: 10.1038/nmeth.2472. Epub 2013 May 5.
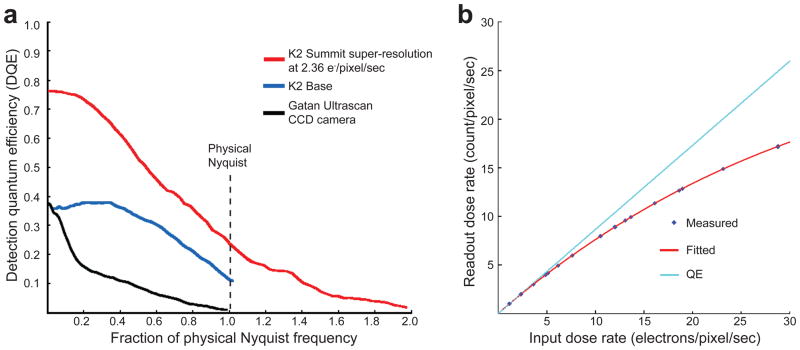
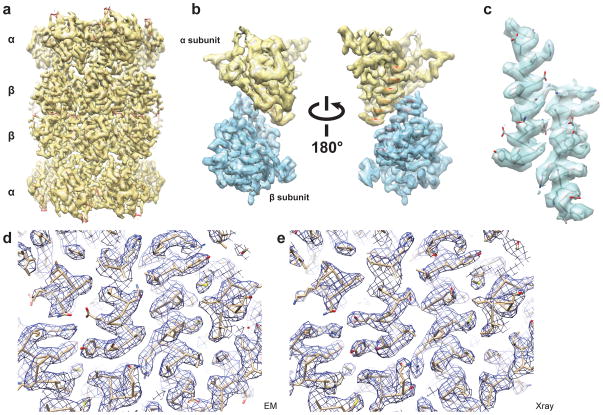
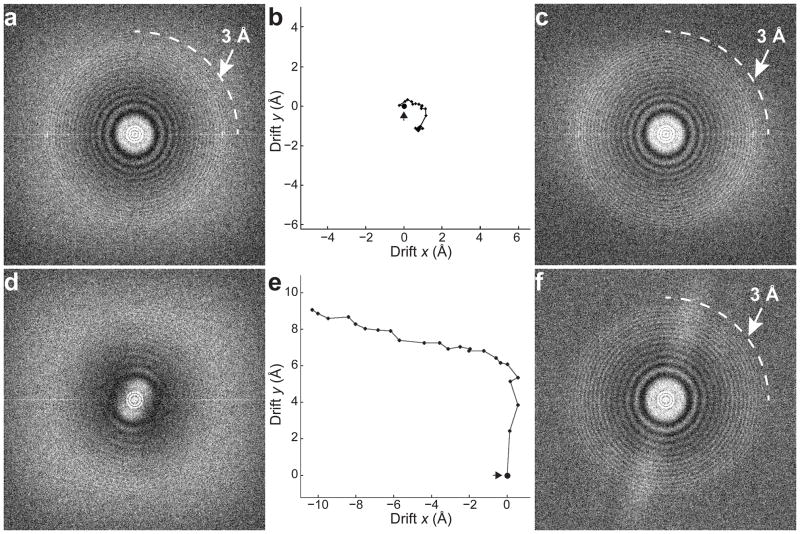
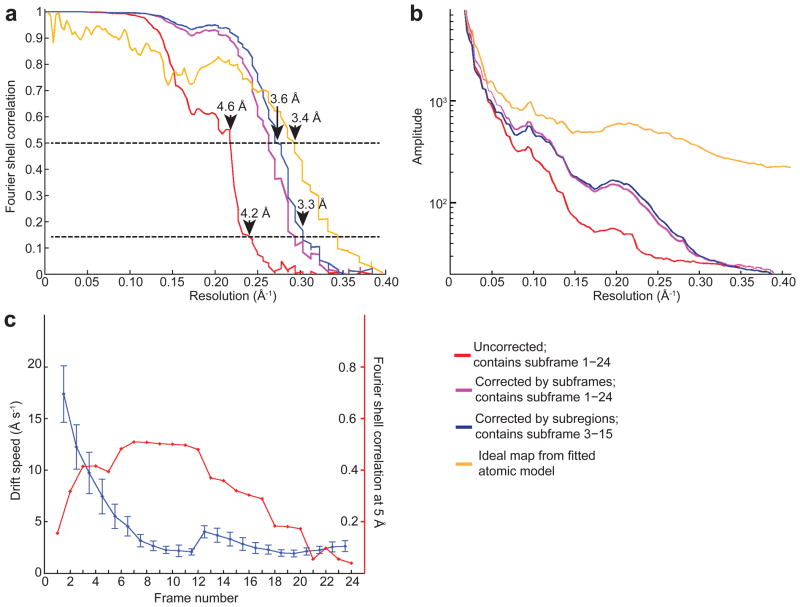
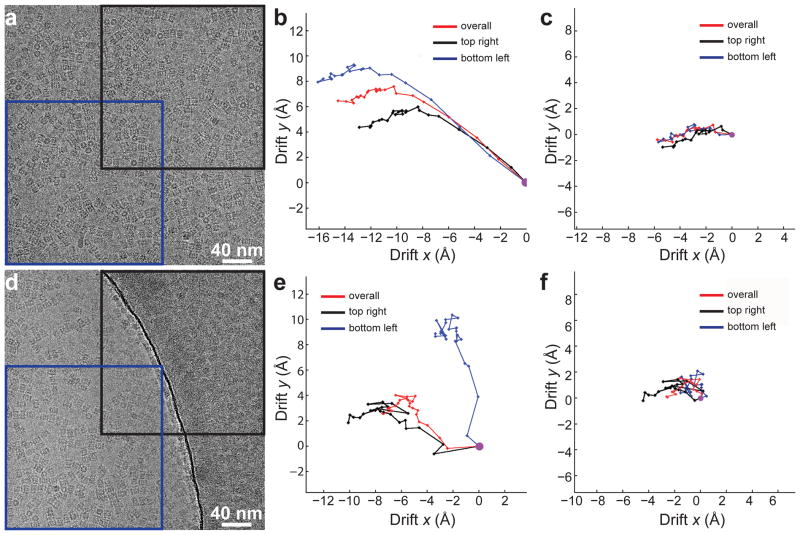
In recent work with large high-symmetry viruses, single-particle electron cryomicroscopy (cryo-EM) has achieved the determination of near-atomic-resolution structures by allowing direct fitting of atomic models into experimental density maps. However, achieving this goal with smaller particles of lower symmetry remains challenging. Using a newly developed single electron-counting detector, we confirmed that electron beam-induced motion substantially degrades resolution, and we showed that the combination of rapid readout and nearly noiseless electron counting allow image blurring to be corrected to subpixel accuracy, restoring intrinsic image information to high resolution (Thon rings visible to ∼3 Å). Using this approach, we determined a 3.3-Å-resolution structure of an ∼700-kDa protein with D7 symmetry, the Thermoplasma acidophilum 20S proteasome, showing clear side-chain density. Our method greatly enhances image quality and data acquisition efficiency-key bottlenecks in applying near-atomic-resolution cryo-EM to a broad range of protein samples.
在最近对大型高对称病毒的研究中,通过将原子模型直接拟合到实验密度图中,单颗粒电子冷冻透射显微镜(cryo-EM)已经实现了近原子分辨率结构的测定。然而,对于较小的、对称性较低的颗粒,实现这一目标仍然具有挑战性。使用新开发的单电子计数探测器,我们证实电子束诱导的运动大大降低了分辨率,并且我们表明,快速读出和几乎无噪声的电子计数的结合允许将图像模糊校正到亚像素精度,从而将内在的图像信息恢复到高分辨率(可见到 ∼3 Å 的径环)。使用这种方法,我们确定了具有 D7 对称性的约 700 kDa 蛋白质的 3.3 Å 分辨率结构,即嗜酸性热原体 20S 蛋白酶体,显示出清晰的侧链密度。我们的方法极大地提高了图像质量和数据采集效率,这是将近原子分辨率 cryo-EM 应用于广泛的蛋白质样品的关键瓶颈。