Li Lun, Long Yan, Zhang Libin, Dalton-Morgan Jessica, Batley Jacqueline, Yu Longjiang, Meng Jinling, Li Maoteng
College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, China; Hubei Bioinformatics and Molecular Imaging Key Laboratory, Huazhong University of Science and Technology, Wuhan, China.
National Key Lab of Crop Genetic Improvement, Huazhong Agricultural University, Wuhan, China; Biotechnology Research Institute, Chinese Academy of Agricultural Sciences, Beijing, China.
PLoS One. 2015 Mar 19;10(3):e0119425. doi: 10.1371/journal.pone.0119425. eCollection 2015.
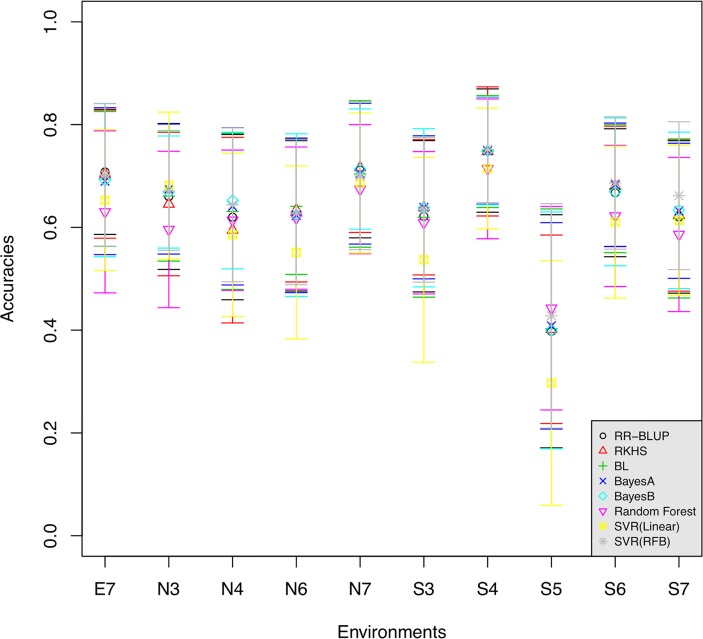
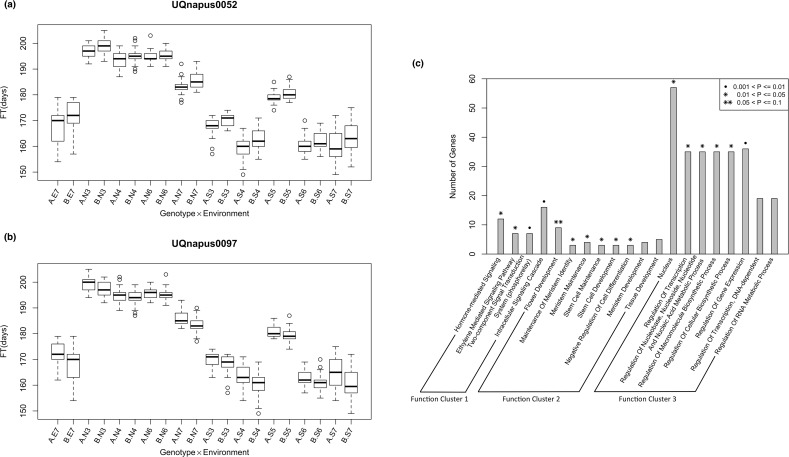
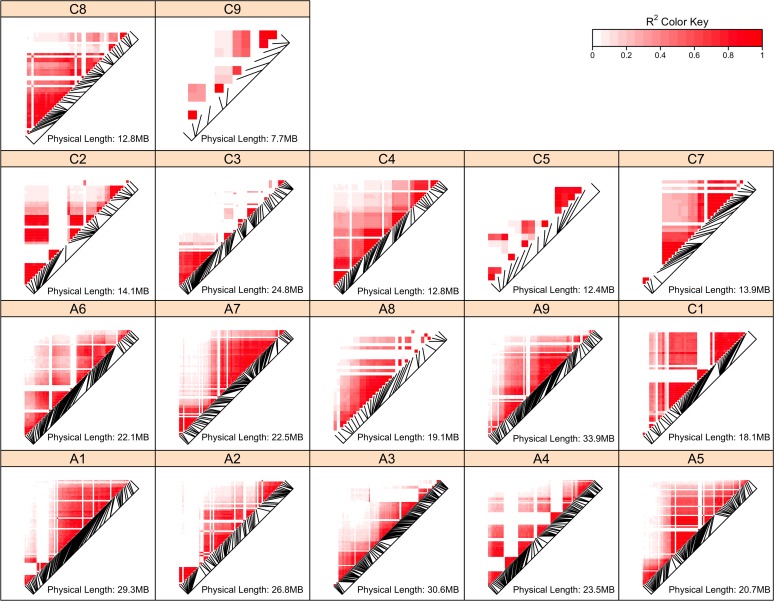
The prediction of the flowering time (FT) trait in Brassica napus based on genome-wide markers and the detection of underlying genetic factors is important not only for oilseed producers around the world but also for the other crop industry in the rotation system in China. In previous studies the low density and mixture of biomarkers used obstructed genomic selection in B. napus and comprehensive mapping of FT related loci. In this study, a high-density genome-wide SNP set was genotyped from a double-haploid population of B. napus. We first performed genomic prediction of FT traits in B. napus using SNPs across the genome under ten environments of three geographic regions via eight existing genomic predictive models. The results showed that all the models achieved comparably high accuracies, verifying the feasibility of genomic prediction in B. napus. Next, we performed a large-scale mapping of FT related loci among three regions, and found 437 associated SNPs, some of which represented known FT genes, such as AP1 and PHYE. The genes tagged by the associated SNPs were enriched in biological processes involved in the formation of flowers. Epistasis analysis showed that significant interactions were found between detected loci, even among some known FT related genes. All the results showed that our large scale and high-density genotype data are of great practical and scientific values for B. napus. To our best knowledge, this is the first evaluation of genomic selection models in B. napus based on a high-density SNP dataset and large-scale mapping of FT loci.
基于全基因组标记预测甘蓝型油菜的开花时间(FT)性状以及检测潜在遗传因素,不仅对全球油菜籽生产者很重要,对中国轮作系统中的其他作物产业也很重要。在以往研究中,所使用生物标志物的低密度和混杂性阻碍了甘蓝型油菜的基因组选择以及FT相关位点的全面定位。在本研究中,从甘蓝型油菜的一个双单倍体群体中对一个高密度全基因组SNP集进行了基因分型。我们首先通过八个现有的基因组预测模型,在三个地理区域的十种环境下,利用全基因组的SNP对甘蓝型油菜的FT性状进行了基因组预测。结果表明,所有模型都达到了相当高的准确性,验证了甘蓝型油菜基因组预测的可行性。接下来,我们在三个区域间对FT相关位点进行了大规模定位,发现了437个相关SNP,其中一些代表已知的FT基因,如AP1和PHYE。由相关SNP标记的基因在花形成所涉及的生物学过程中富集。上位性分析表明,在检测到的位点之间发现了显著的相互作用,甚至在一些已知的FT相关基因之间也是如此。所有结果表明,我们的大规模和高密度基因型数据对甘蓝型油菜具有很大的实践和科学价值。据我们所知,这是首次基于高密度SNP数据集和FT位点的大规模定位对甘蓝型油菜的基因组选择模型进行评估。