Obenauf Anna C, Zou Yilong, Ji Andrew L, Vanharanta Sakari, Shu Weiping, Shi Hubing, Kong Xiangju, Bosenberg Marcus C, Wiesner Thomas, Rosen Neal, Lo Roger S, Massagué Joan
Cancer Biology and Genetics Program, Memorial Sloan Kettering Cancer Center, New York, New York 10065, USA.
1] Cancer Biology and Genetics Program, Memorial Sloan Kettering Cancer Center, New York, New York 10065, USA [2] Gerstner Sloan Kettering School of Biomedical Sciences, Memorial Sloan Kettering Cancer Center, New York, New York 10065, USA.
Nature. 2015 Apr 16;520(7547):368-72. doi: 10.1038/nature14336. Epub 2015 Mar 25.
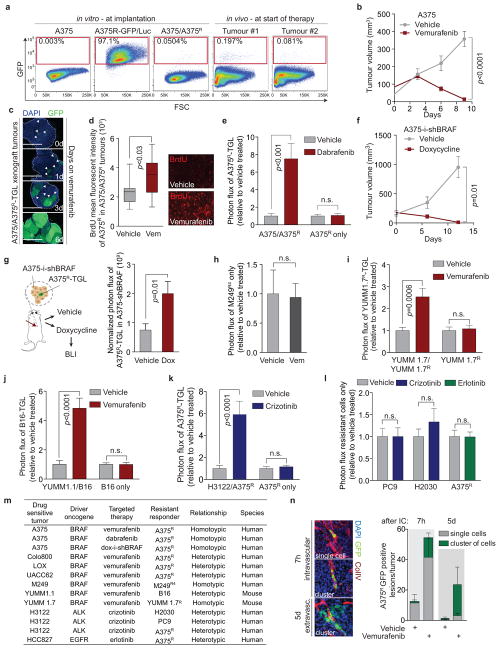
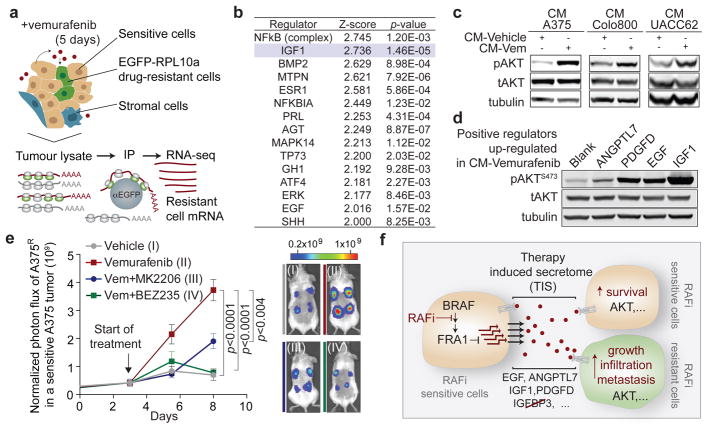
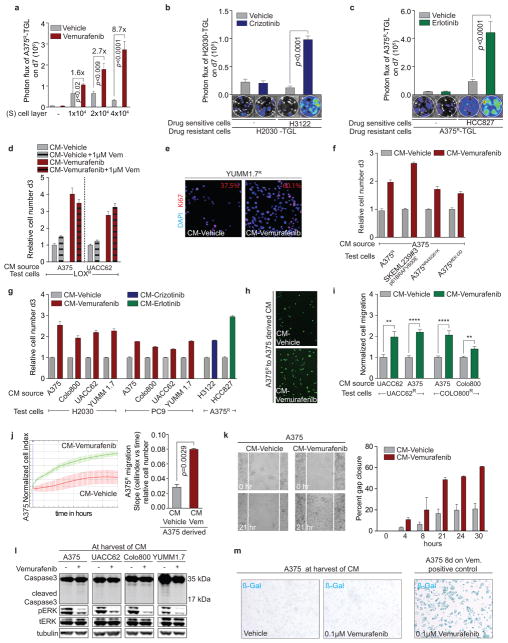
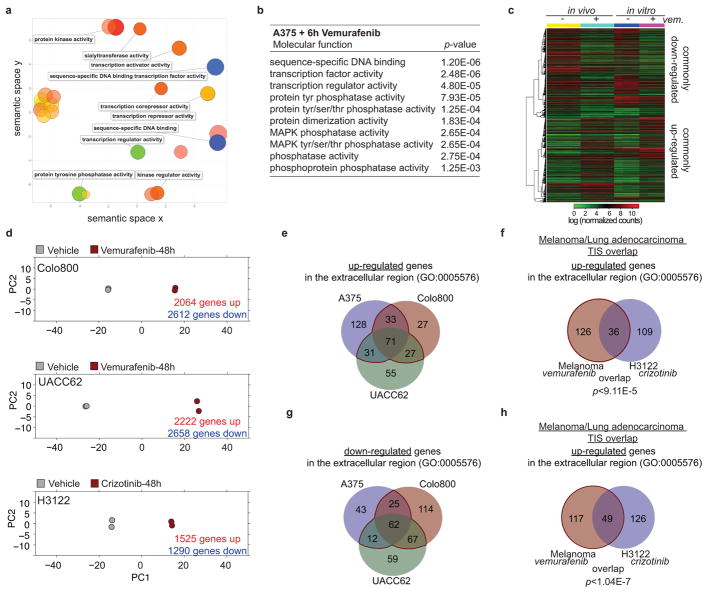
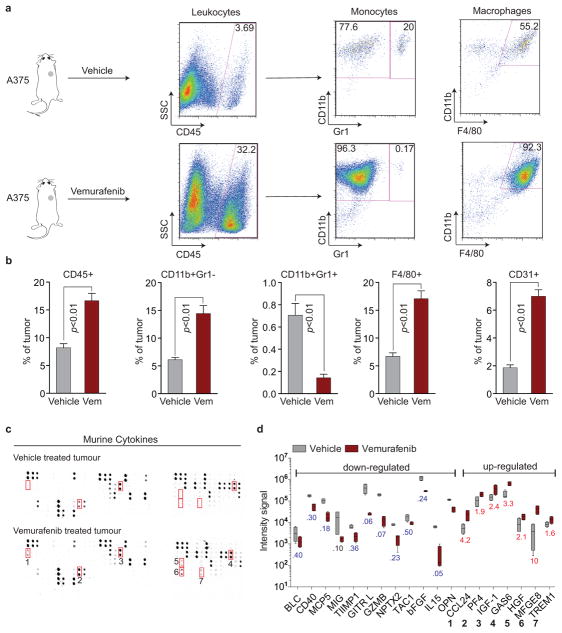
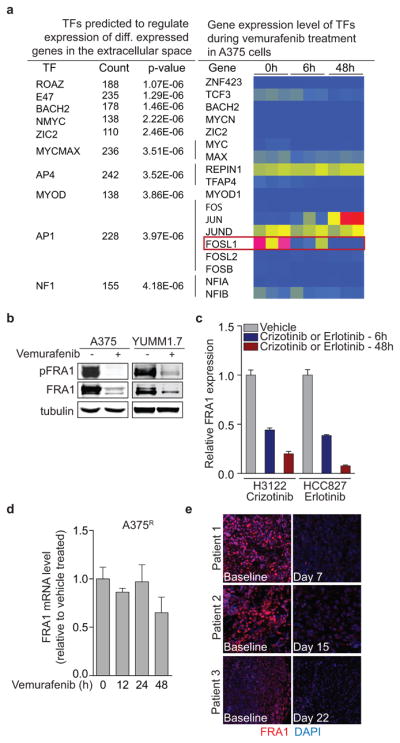
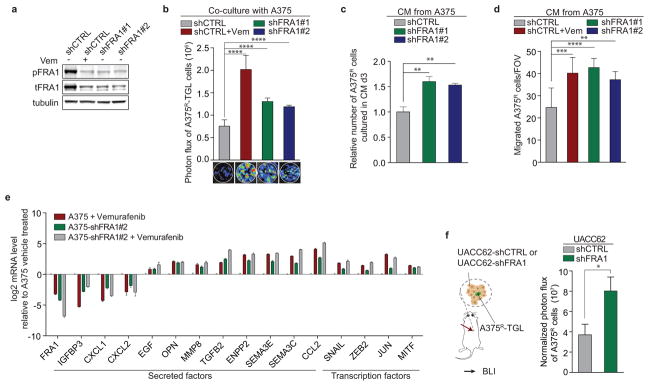
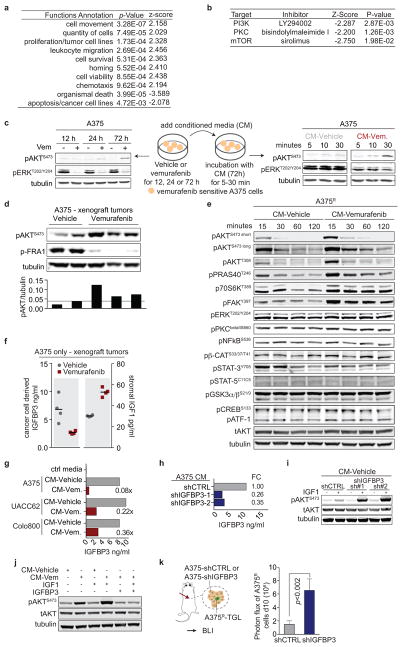
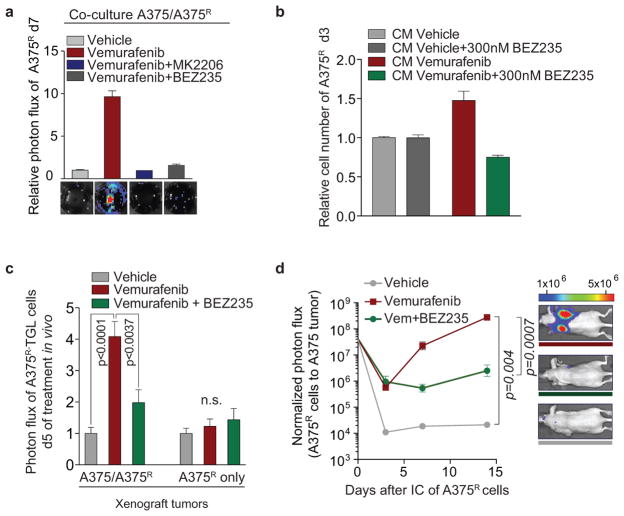
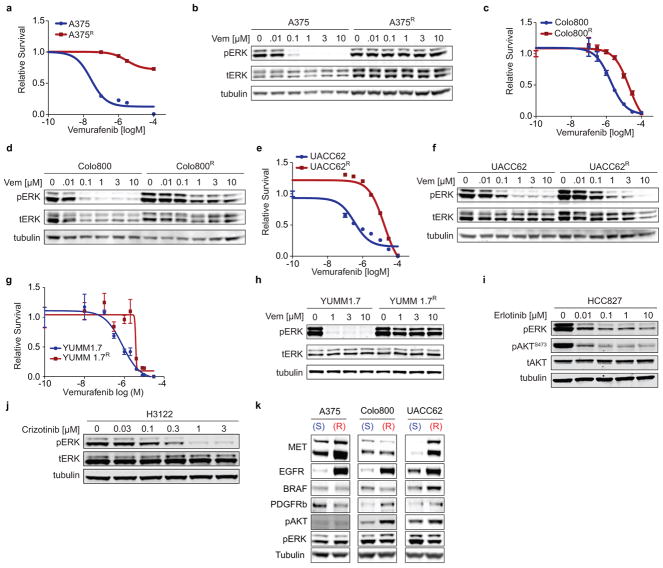
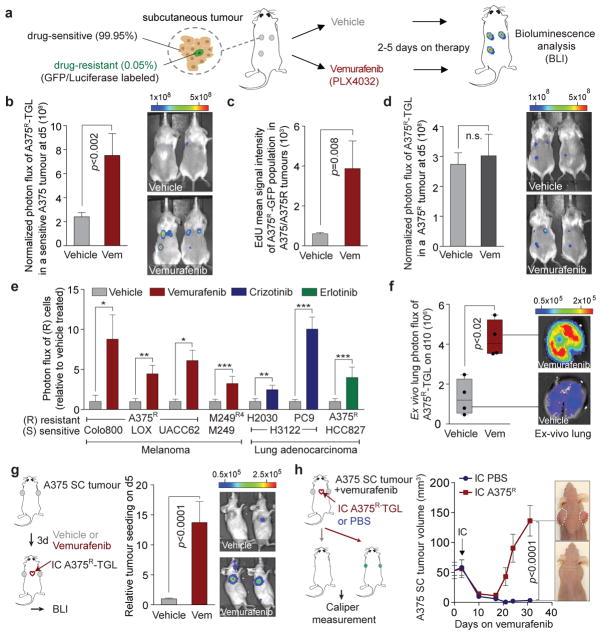
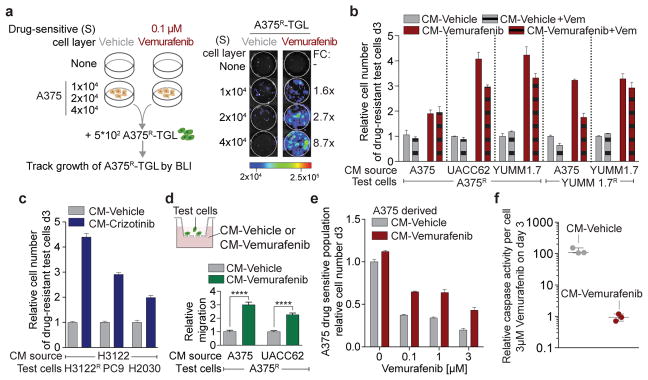
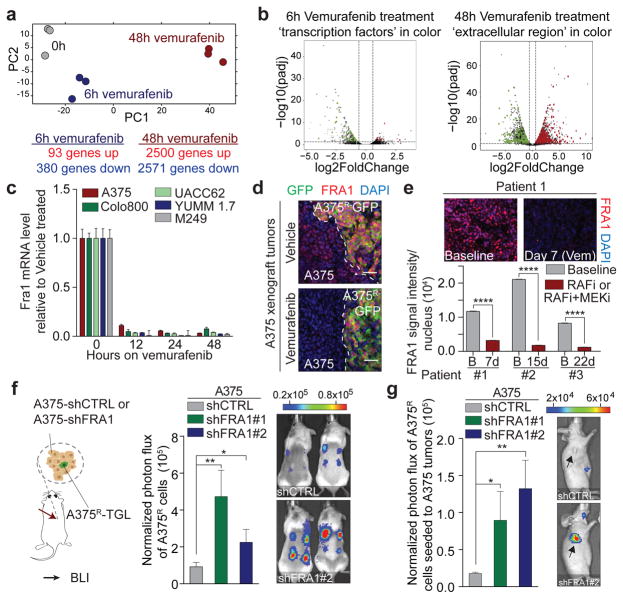
Drug resistance invariably limits the clinical efficacy of targeted therapy with kinase inhibitors against cancer. Here we show that targeted therapy with BRAF, ALK or EGFR kinase inhibitors induces a complex network of secreted signals in drug-stressed human and mouse melanoma and human lung adenocarcinoma cells. This therapy-induced secretome stimulates the outgrowth, dissemination and metastasis of drug-resistant cancer cell clones and supports the survival of drug-sensitive cancer cells, contributing to incomplete tumour regression. The tumour-promoting secretome of melanoma cells treated with the kinase inhibitor vemurafenib is driven by downregulation of the transcription factor FRA1. In situ transcriptome analysis of drug-resistant melanoma cells responding to the regressing tumour microenvironment revealed hyperactivation of several signalling pathways, most prominently the AKT pathway. Dual inhibition of RAF and the PI(3)K/AKT/mTOR intracellular signalling pathways blunted the outgrowth of the drug-resistant cell population in BRAF mutant human melanoma, suggesting this combination therapy as a strategy against tumour relapse. Thus, therapeutic inhibition of oncogenic drivers induces vast secretome changes in drug-sensitive cancer cells, paradoxically establishing a tumour microenvironment that supports the expansion of drug-resistant clones, but is susceptible to combination therapy.
耐药性总是会限制激酶抑制剂靶向治疗癌症的临床疗效。在此,我们表明,使用BRAF、ALK或EGFR激酶抑制剂进行靶向治疗会在药物应激的人源和鼠源黑色素瘤细胞以及人肺腺癌细胞中诱导出一个复杂的分泌信号网络。这种治疗诱导的分泌组会刺激耐药癌细胞克隆的生长、扩散和转移,并支持药物敏感癌细胞的存活,导致肿瘤不完全消退。用激酶抑制剂维莫非尼治疗的黑色素瘤细胞的促肿瘤分泌组是由转录因子FRA1的下调驱动的。对响应肿瘤消退微环境的耐药黑色素瘤细胞进行原位转录组分析发现,几种信号通路高度激活,最显著的是AKT通路。对RAF和PI(3)K/AKT/mTOR细胞内信号通路的双重抑制减弱了BRAF突变型人黑色素瘤中耐药细胞群体的生长,表明这种联合治疗可作为一种对抗肿瘤复发的策略。因此,对致癌驱动因子的治疗性抑制会在药物敏感癌细胞中引起大量分泌组变化,反常地建立起一个支持耐药克隆扩增但易受联合治疗影响的肿瘤微环境。