Zhang Pei-Wei, Chen Lei, Huang Tao, Zhang Ning, Kong Xiang-Yin, Cai Yu-Dong
The Key Laboratory of Stem Cell Biology, Institute of Health Sciences, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, Shanghai, P.R. China.
College of Information Engineering, Shanghai Maritime University, Shanghai, P.R. China.
PLoS One. 2015 Mar 30;10(3):e0123147. doi: 10.1371/journal.pone.0123147. eCollection 2015.
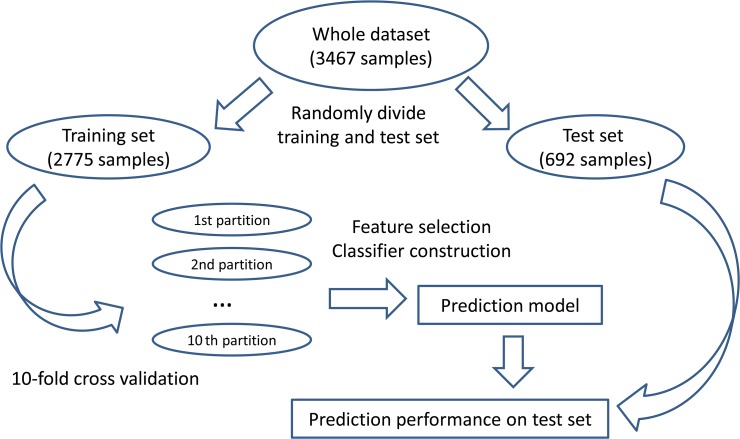
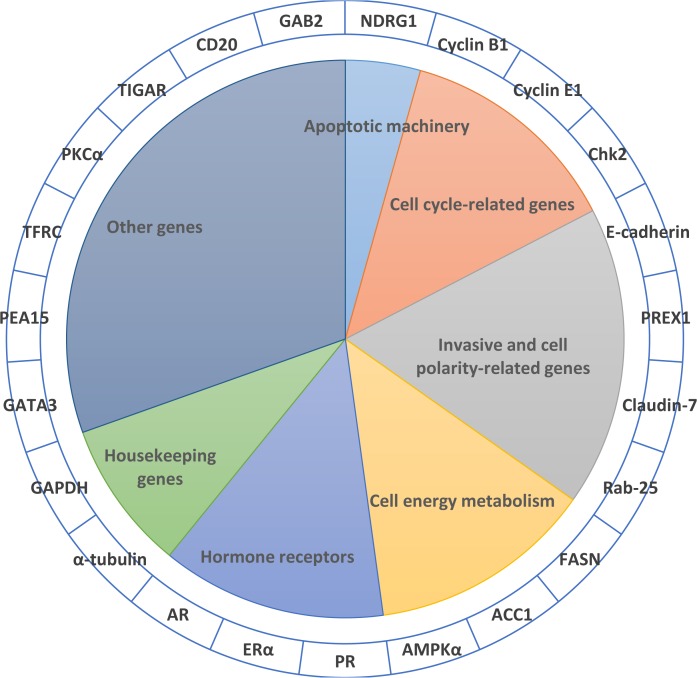
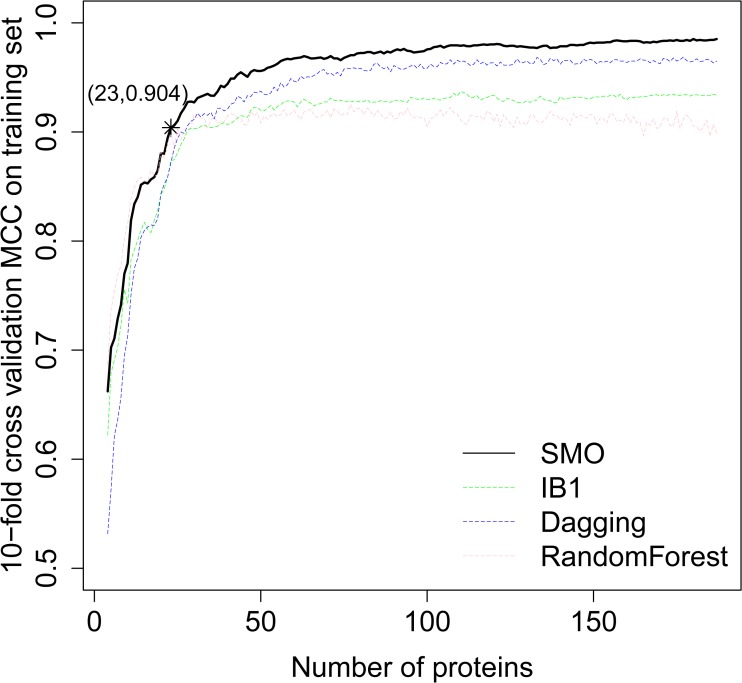
Gathering vast data sets of cancer genomes requires more efficient and autonomous procedures to classify cancer types and to discover a few essential genes to distinguish different cancers. Because protein expression is more stable than gene expression, we chose reverse phase protein array (RPPA) data, a powerful and robust antibody-based high-throughput approach for targeted proteomics, to perform our research. In this study, we proposed a computational framework to classify the patient samples into ten major cancer types based on the RPPA data using the SMO (Sequential minimal optimization) method. A careful feature selection procedure was employed to select 23 important proteins from the total of 187 proteins by mRMR (minimum Redundancy Maximum Relevance Feature Selection) and IFS (Incremental Feature Selection) on the training set. By using the 23 proteins, we successfully classified the ten cancer types with an MCC (Matthews Correlation Coefficient) of 0.904 on the training set, evaluated by 10-fold cross-validation, and an MCC of 0.936 on an independent test set. Further analysis of these 23 proteins was performed. Most of these proteins can present the hallmarks of cancer; Chk2, for example, plays an important role in the proliferation of cancer cells. Our analysis of these 23 proteins lends credence to the importance of these genes as indicators of cancer classification. We also believe our methods and findings may shed light on the discoveries of specific biomarkers of different types of cancers.
收集大量癌症基因组数据集需要更高效、自主的程序来对癌症类型进行分类,并发现一些区分不同癌症的关键基因。由于蛋白质表达比基因表达更稳定,我们选择了反相蛋白质阵列(RPPA)数据,这是一种基于抗体的强大且稳健的靶向蛋白质组学高通量方法,来开展我们的研究。在本研究中,我们提出了一个计算框架,使用SMO(序列最小优化)方法基于RPPA数据将患者样本分类为十种主要癌症类型。通过在训练集上使用mRMR(最小冗余最大相关特征选择)和IFS(增量特征选择),我们采用了仔细的特征选择程序从总共187种蛋白质中选择了23种重要蛋白质。使用这23种蛋白质,我们在训练集上通过10折交叉验证成功将十种癌症类型分类,马修斯相关系数(MCC)为0.904,在独立测试集上的MCC为0.936。对这23种蛋白质进行了进一步分析。这些蛋白质中的大多数都能呈现癌症的特征;例如,Chk2在癌细胞增殖中起重要作用。我们对这23种蛋白质的分析证实了这些基因作为癌症分类指标的重要性。我们也相信我们的方法和发现可能会为不同类型癌症的特定生物标志物的发现提供线索。