De Spiegelaere Ward, Dern-Wieloch Jutta, Weigel Roswitha, Schumacher Valérie, Schorle Hubert, Nettersheim Daniel, Bergmann Martin, Brehm Ralph, Kliesch Sabine, Vandekerckhove Linos, Fink Cornelia
Ghent University, Department of Internal Medicine, Ghent, Belgium.
Justus-Liebig-University, Department of Veterinary Anatomy, Histology and Embryology, Giessen, Germany.
PLoS One. 2015 Mar 31;10(3):e0122515. doi: 10.1371/journal.pone.0122515. eCollection 2015.
An appropriate normalization strategy is crucial for data analysis from real time reverse transcription polymerase chain reactions (RT-qPCR). It is widely supported to identify and validate stable reference genes, since no single biological gene is stably expressed between cell types or within cells under different conditions. Different algorithms exist to validate optimal reference genes for normalization. Applying human cells, we here compare the three main methods to the online available RefFinder tool that integrates these algorithms along with R-based software packages which include the NormFinder and GeNorm algorithms.
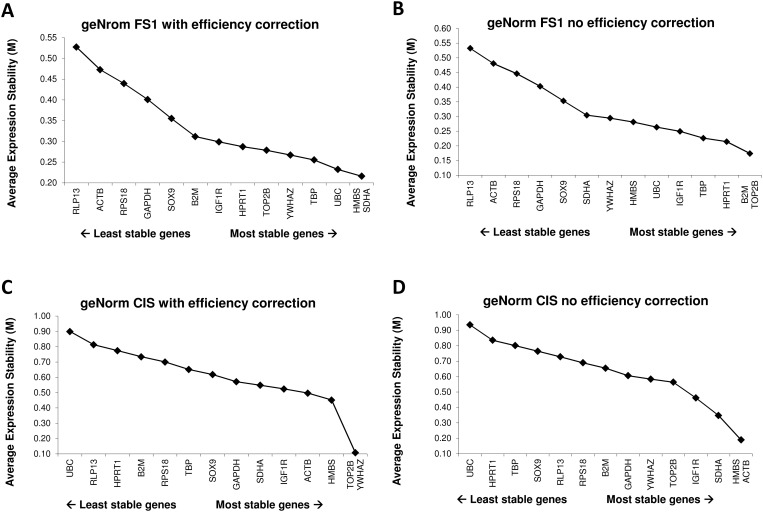
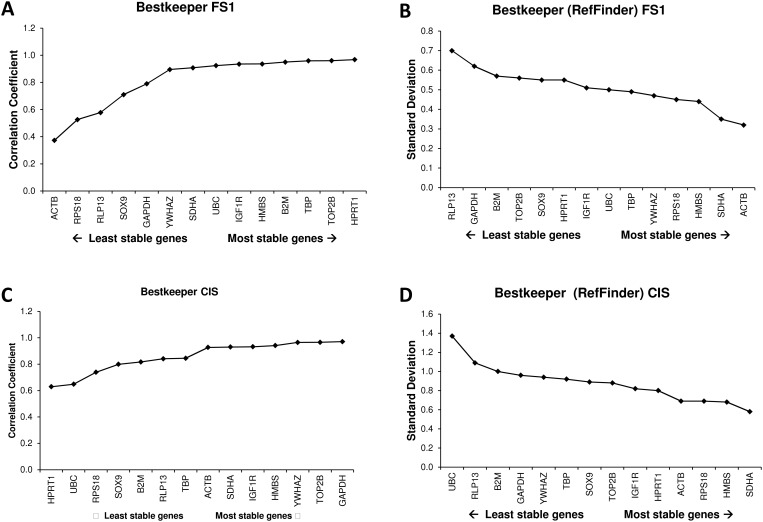
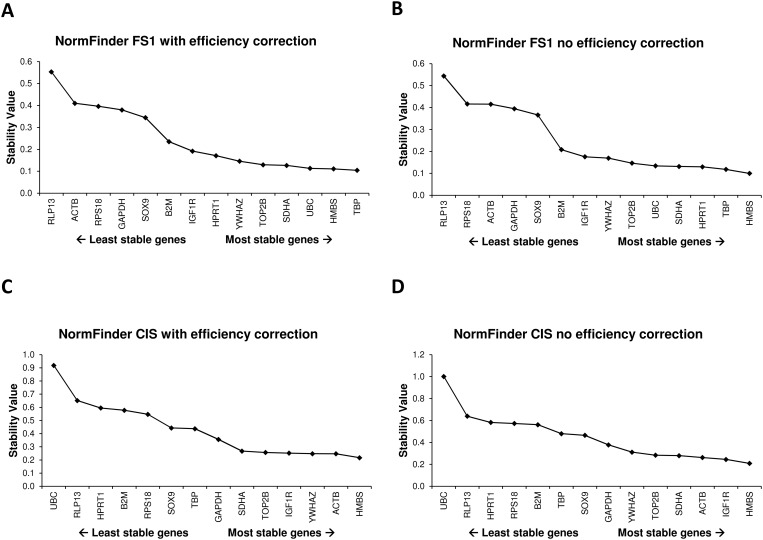
14 candidate reference genes were assessed by RT-qPCR in two sample sets, i.e. a set of samples of human testicular tissue containing carcinoma in situ (CIS), and a set of samples from the human adult Sertoli cell line (FS1) either cultured alone or in co-culture with the seminoma like cell line (TCam-2) or with equine bone marrow derived mesenchymal stem cells (eBM-MSC). Expression stabilities of the reference genes were evaluated using geNorm, NormFinder, and BestKeeper. Similar results were obtained by the three approaches for the most and least stably expressed genes. The R-based packages NormqPCR, SLqPCR and the NormFinder for R script gave identical gene rankings. Interestingly, different outputs were obtained between the original software packages and the RefFinder tool, which is based on raw Cq values for input. When the raw data were reanalysed assuming 100% efficiency for all genes, then the outputs of the original software packages were similar to the RefFinder software, indicating that RefFinder outputs may be biased because PCR efficiencies are not taken into account.
This report shows that assay efficiency is an important parameter for reference gene validation. New software tools that incorporate these algorithms should be carefully validated prior to use.
合适的标准化策略对于实时逆转录聚合酶链反应(RT-qPCR)的数据分析至关重要。由于在不同细胞类型之间或同一细胞在不同条件下,没有单一的生物基因能稳定表达,因此识别和验证稳定的内参基因得到了广泛支持。存在不同的算法来验证用于标准化的最佳内参基因。我们使用人类细胞,将三种主要方法与在线可用的RefFinder工具进行比较,该工具整合了这些算法以及基于R的软件包,其中包括NormFinder和GeNorm算法。
通过RT-qPCR在两个样本集中评估了14个候选内参基因,即一组包含原位癌(CIS)的人类睾丸组织样本,以及一组来自人类成年支持细胞系(FS1)的样本,这些样本要么单独培养,要么与精原细胞瘤样细胞系(TCam-2)或马骨髓来源的间充质干细胞(eBM-MSC)共培养。使用geNorm、NormFinder和BestKeeper评估内参基因的表达稳定性。对于表达最稳定和最不稳定的基因,这三种方法获得了相似的结果。基于R的软件包NormqPCR、SLqPCR和R脚本的NormFinder给出了相同的基因排名。有趣的是,原始软件包和基于原始Cq值输入的RefFinder工具之间获得了不同的输出。当假设所有基因的效率为100%对原始数据重新分析时,那么原始软件包的输出与RefFinder软件相似,这表明RefFinder的输出可能存在偏差,因为未考虑PCR效率。
本报告表明检测效率是内参基因验证的一个重要参数。在使用之前,应仔细验证纳入这些算法的新软件工具。