Kay Gemma L, Sergeant Martin J, Zhou Zhemin, Chan Jacqueline Z-M, Millard Andrew, Quick Joshua, Szikossy Ildikó, Pap Ildikó, Spigelman Mark, Loman Nicholas J, Achtman Mark, Donoghue Helen D, Pallen Mark J
Microbiology and Infection Unit, Division of Translational and Systems Medicine, Warwick Medical School, University of Warwick, Gibbet Hill Road, Coventry CV4 7AL, UK.
Institute of Microbiology and Infection, School of Biosciences, University of Birmingham, Birmingham B15 2TT, UK.
Nat Commun. 2015 Apr 7;6:6717. doi: 10.1038/ncomms7717.
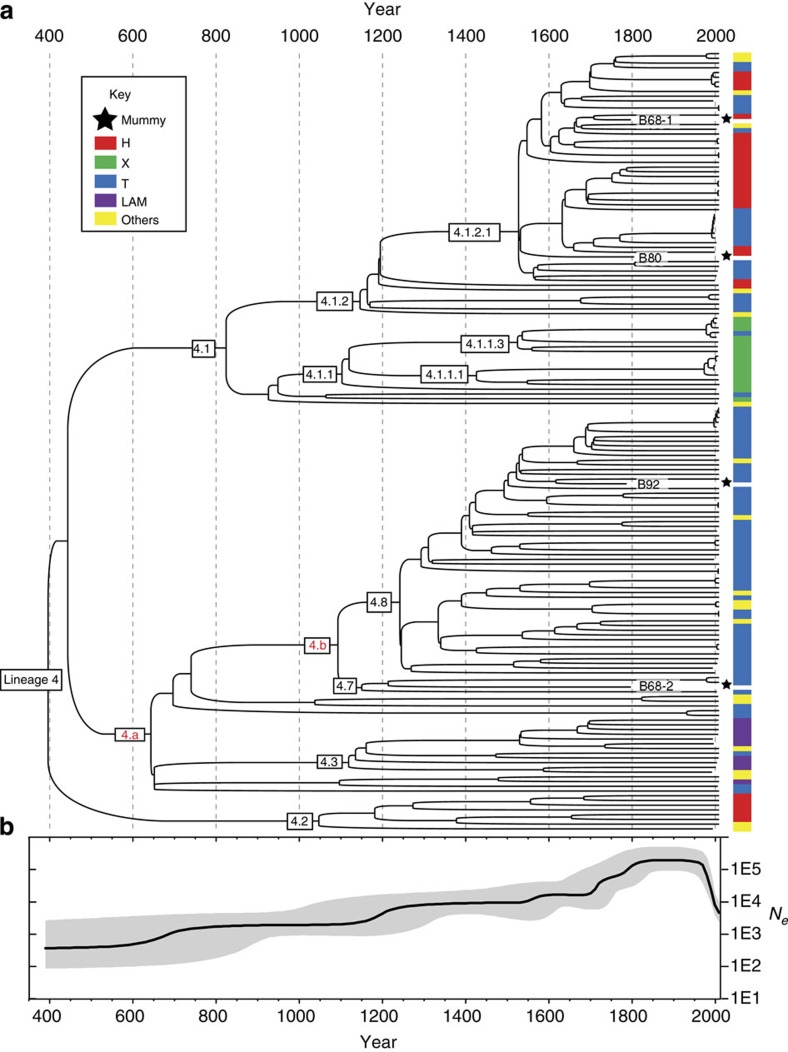
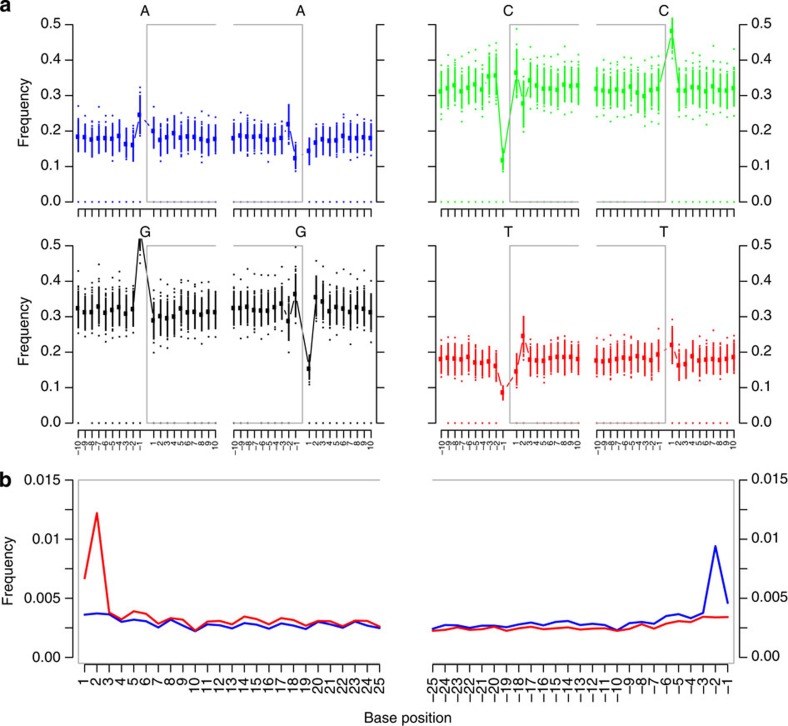
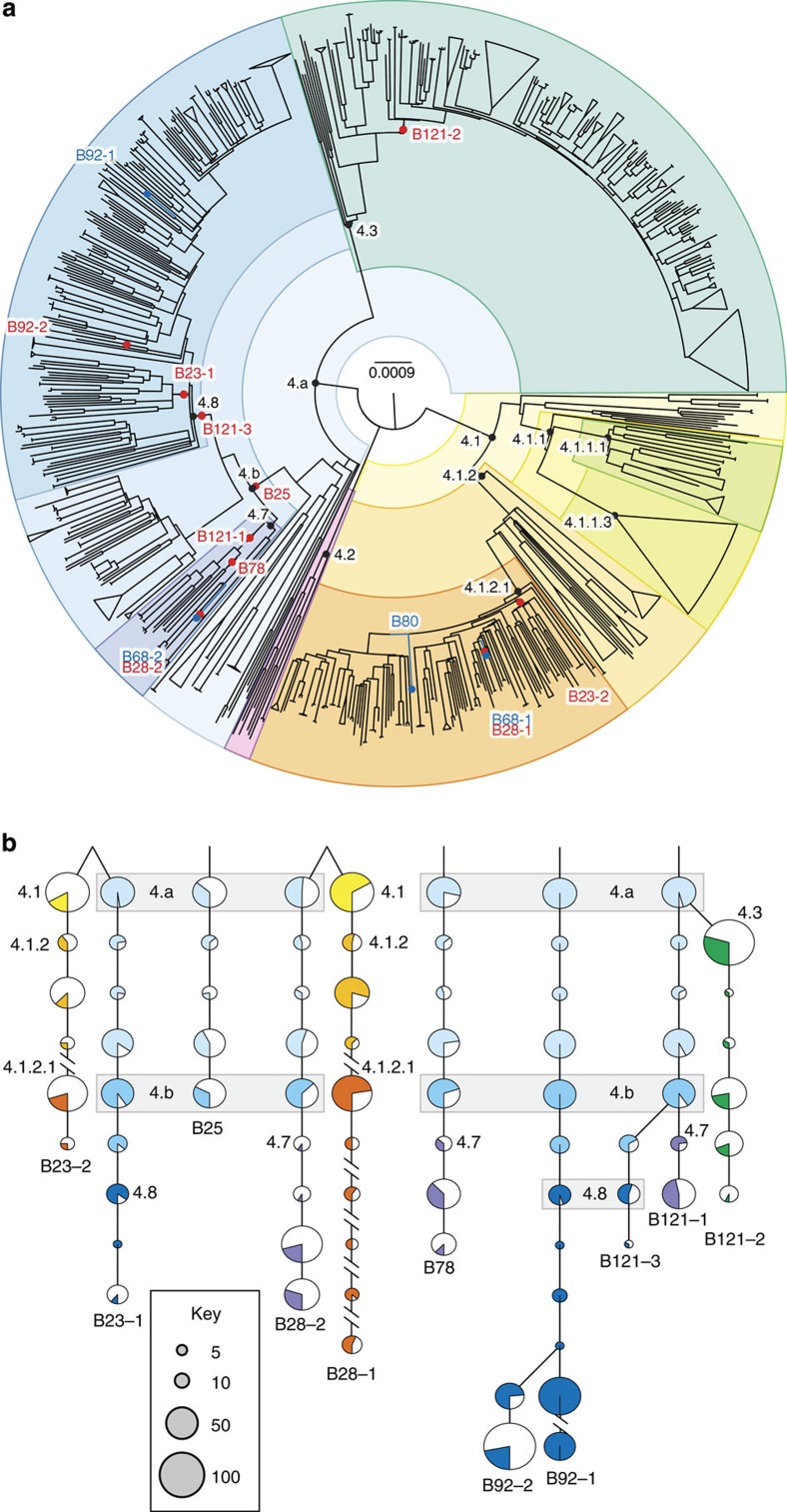
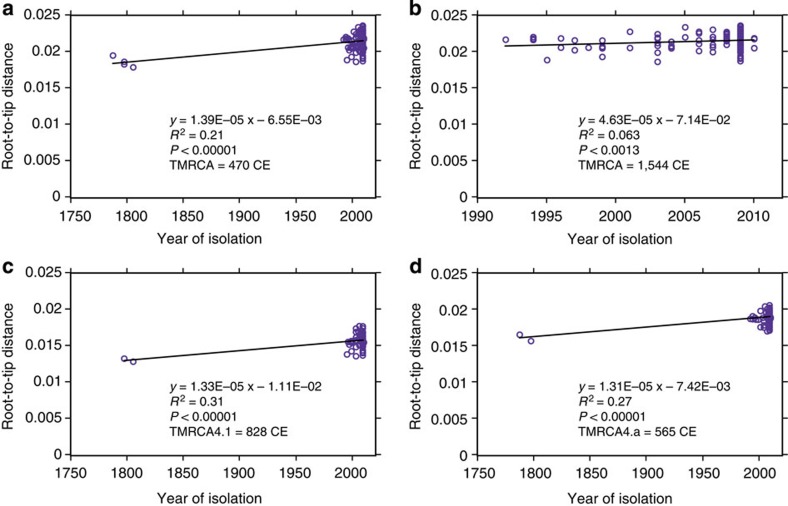
Tuberculosis (TB) was once a major killer in Europe, but it is unclear how the strains and patterns of infection at 'peak TB' relate to what we see today. Here we describe 14 genome sequences of M. tuberculosis, representing 12 distinct genotypes, obtained from human remains from eighteenth-century Hungary using metagenomics. All our historic genotypes belong to M. tuberculosis Lineage 4. Bayesian phylogenetic dating, based on samples with well-documented dates, places the most recent common ancestor of this lineage in the late Roman period. We find that most bodies yielded more than one M. tuberculosis genotype and we document an intimate epidemiological link between infections in two long-dead individuals. Our results suggest that metagenomic approaches usefully inform detection and characterization of historical and contemporary infections.
结核病曾是欧洲的主要杀手,但“结核病高峰期”的感染菌株和模式与我们如今所见的情况之间的关系尚不清楚。在此,我们描述了从18世纪匈牙利人类遗骸中利用宏基因组学获得的14个结核分枝杆菌基因组序列,代表12种不同的基因型。我们所有的历史基因型均属于结核分枝杆菌谱系4。基于日期记录完备的样本进行的贝叶斯系统发育年代测定将该谱系的最近共同祖先置于罗马晚期。我们发现大多数尸体产生了不止一种结核分枝杆菌基因型,并且我们记录了两个早已去世个体感染之间密切的流行病学联系。我们的结果表明,宏基因组学方法有助于对历史和当代感染进行检测和特征描述。