Larkins Nicholas, Wallis Mathew, McGillivray Barbara, Mammen Cherry
Division of Nephrology, Department of Pediatrics , University of British Columbia , Vancouver , Canada.
Department of Medical Genetics , Sydney Children's Hospital , Sydney , Australia.
Clin Kidney J. 2014 Jun;7(3):306-10. doi: 10.1093/ckj/sfu029. Epub 2014 Apr 4.
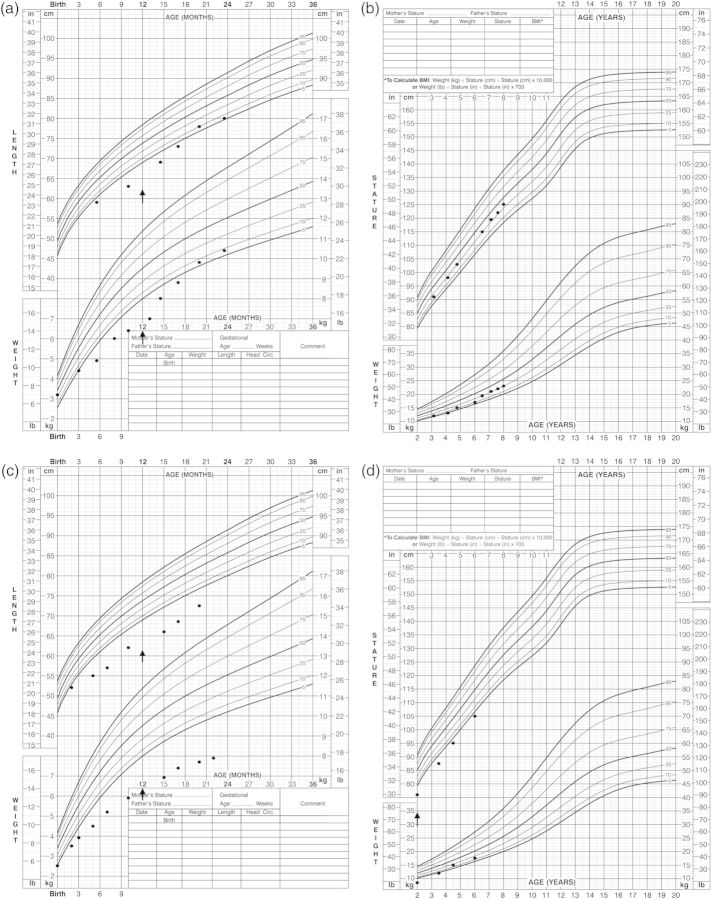
Our understanding of Gitelman syndrome (GS) and Bartter syndrome has continued to evolve with the use of genetic testing to more precisely define the tubular defects responsible. GS is caused by mutations in the SLC12A3 gene encoding the Na(+)-Cl(-) co-transporter of the distal convoluted tubule (NCCT) and tends to be associated with a milder salt-losing phenotype. We describe two female siblings presenting in infancy with a severe salt-losing tubulopathy and failure to thrive due to compound heterozygous mutations in the SLC12A3 gene encoding the NCCT. Both children were treated with indomethacin resulting in improved linear growth and polyuria. Some atypical biochemical findings in our cases are discussed including raised urinary prostaglandin (PGE2) excretion that normalized with intravenous fluid repletion.
随着基因检测的应用,我们对吉特曼综合征(GS)和巴特综合征的认识不断发展,从而能更精确地确定导致肾小管缺陷的原因。GS由编码远曲小管钠氯共转运体(NCCT)的SLC12A3基因突变引起,往往与较轻的失盐表型相关。我们描述了两名女性同胞婴儿,她们因编码NCCT的SLC12A3基因复合杂合突变,在婴儿期出现严重的失盐性肾小管病且生长发育不良。两名患儿均接受了吲哚美辛治疗,线性生长和多尿症状得到改善。我们讨论了病例中的一些非典型生化检查结果,包括尿前列腺素(PGE2)排泄增加,经静脉补液后恢复正常。