Chen Siyuan, Ren Chengzhi, Zhai Jingjing, Yu Jiantao, Zhao Xuyang, Li Zelong, Zhang Ting, Ma Wenlong, Han Zhaoxue, Ma Chuang
State Key Laboratory of Crop Stress Biology for Arid Areas, Center of Bioinformatics, College of Life Sciences, Northwest Agriculture and Forestry University.
College of Information Engineering, Northwest Agriculture and Forestry University.
Brief Bioinform. 2020 Mar 23;21(2):676-686. doi: 10.1093/bib/bbz018.
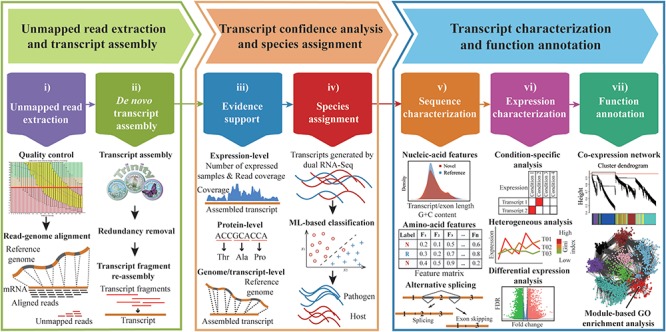
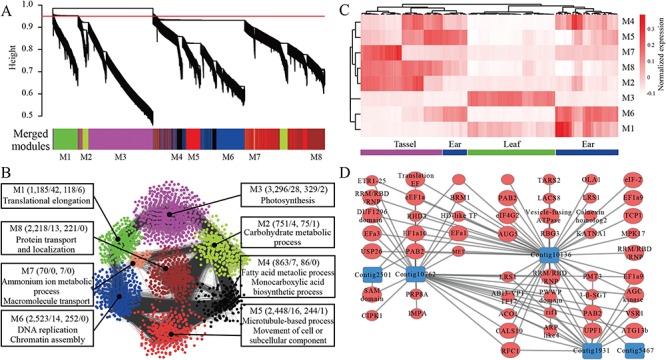
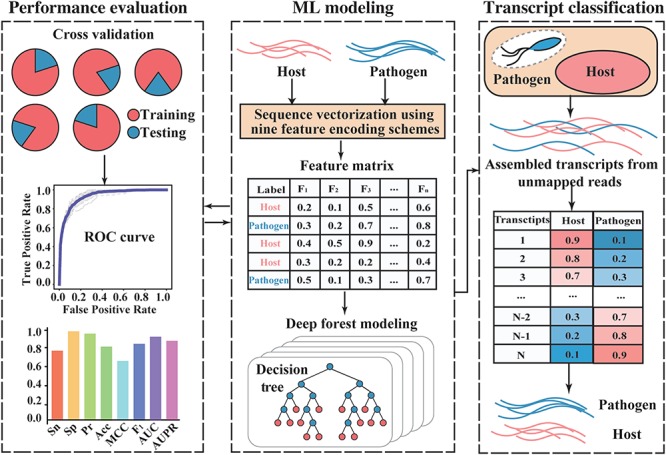
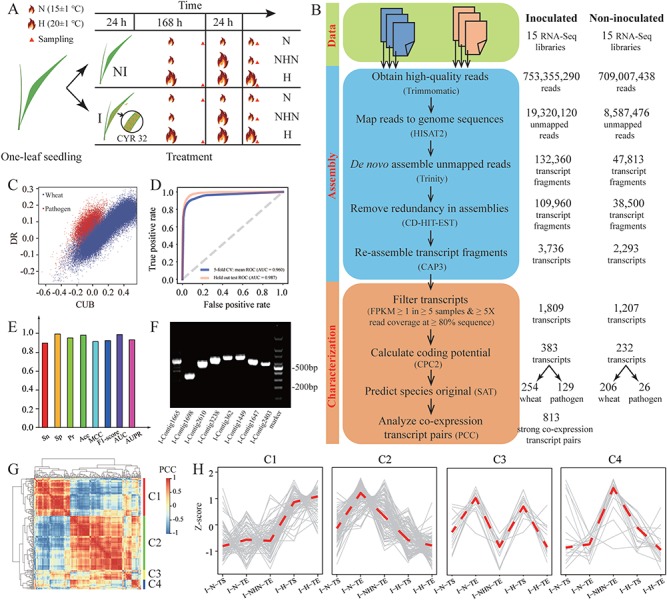
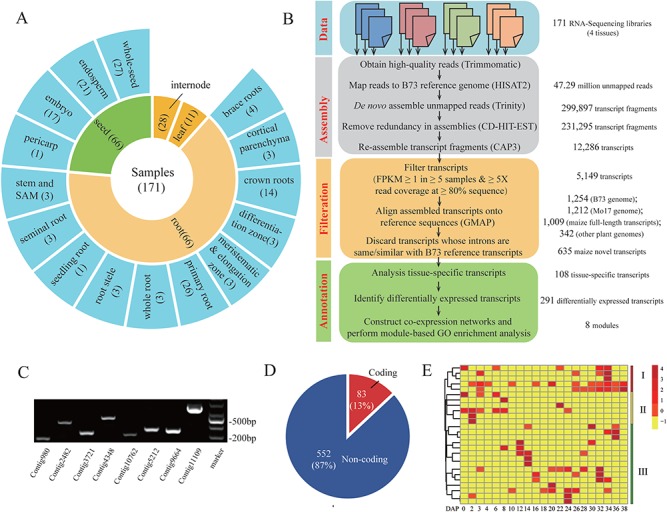
A widely used approach in transcriptome analysis is the alignment of short reads to a reference genome. However, owing to the deficiencies of specially designed analytical systems, short reads unmapped to the genome sequence are usually ignored, resulting in the loss of significant biological information and insights. To fill this gap, we present Comprehensive Assembly and Functional annotation of Unmapped RNA-Seq data (CAFU), a Galaxy-based framework that can facilitate the large-scale analysis of unmapped RNA sequencing (RNA-Seq) reads from single- and mixed-species samples. By taking advantage of machine learning techniques, CAFU addresses the issue of accurately identifying the species origin of transcripts assembled using unmapped reads from mixed-species samples. CAFU also represents an innovation in that it provides a comprehensive collection of functions required for transcript confidence evaluation, coding potential calculation, sequence and expression characterization and function annotation. These functions and their dependencies have been integrated into a Galaxy framework that provides access to CAFU via a user-friendly interface, dramatically simplifying complex exploration tasks involving unmapped RNA-Seq reads. CAFU has been validated with RNA-Seq data sets from wheat and Zea mays (maize) samples. CAFU is freely available via GitHub: https://github.com/cma2015/CAFU.
转录组分析中一种广泛使用的方法是将短读段与参考基因组进行比对。然而,由于专门设计的分析系统存在缺陷,未映射到基因组序列的短读段通常被忽略,导致重要生物学信息和见解的丢失。为了填补这一空白,我们提出了未映射RNA-Seq数据的全面组装和功能注释(CAFU),这是一个基于Galaxy的框架,可促进对来自单物种和混合物种样本的未映射RNA测序(RNA-Seq)读段进行大规模分析。通过利用机器学习技术,CAFU解决了准确识别使用来自混合物种样本的未映射读段组装的转录本的物种来源的问题。CAFU还具有创新性,它提供了转录本置信度评估、编码潜力计算、序列和表达特征分析以及功能注释所需的全面功能集合。这些功能及其依赖关系已集成到一个Galaxy框架中,该框架通过用户友好的界面提供对CAFU的访问,极大地简化了涉及未映射RNA-Seq读段的复杂探索任务。CAFU已通过来自小麦和玉米样本的RNA-Seq数据集进行了验证。可通过GitHub免费获取CAFU:https://github.com/cma2015/CAFU。