Rodríguez-Ramilo Silvia Teresa, Fernández Jesús, Toro Miguel Angel, Hernández Delfino, Villanueva Beatriz
Departamento de Mejora Genética Animal, INIA, Madrid, Spain.
Departamento de Producción Animal, Escuela Técnica Superior de Ingenieros Agrónomos, Madrid, Spain.
PLoS One. 2015 Apr 16;10(4):e0124157. doi: 10.1371/journal.pone.0124157. eCollection 2015.
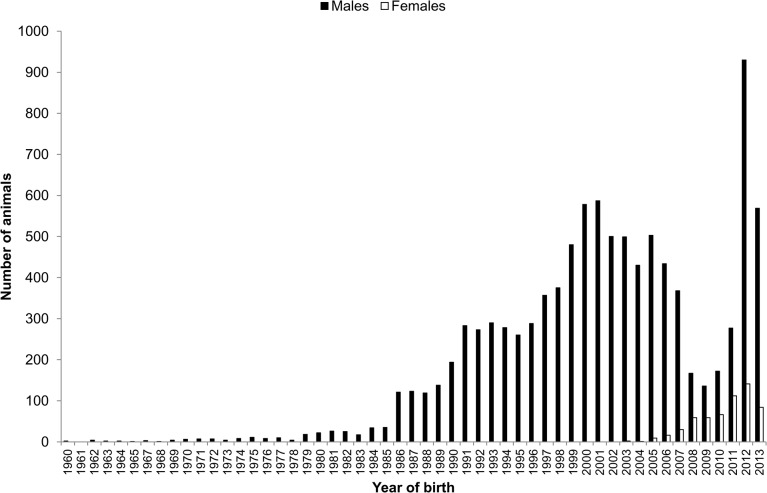
Estimates of effective population size in the Holstein cattle breed have usually been low despite the large number of animals that constitute this breed. Effective population size is inversely related to the rates at which coancestry and inbreeding increase and these rates have been high as a consequence of intense and accurate selection. Traditionally, coancestry and inbreeding coefficients have been calculated from pedigree data. However, the development of genome-wide single nucleotide polymorphisms has increased the interest of calculating these coefficients from molecular data in order to improve their accuracy. In this study, genomic estimates of coancestry, inbreeding and effective population size were obtained in the Spanish Holstein population and then compared with pedigree-based estimates. A total of 11,135 animals genotyped with the Illumina BovineSNP50 BeadChip were available for the study. After applying filtering criteria, the final genomic dataset included 36,693 autosomal SNPs and 10,569 animals. Pedigree data from those genotyped animals included 31,203 animals. These individuals represented only the last five generations in order to homogenise the amount of pedigree information across animals. Genomic estimates of coancestry and inbreeding were obtained from identity by descent segments (coancestry) or runs of homozygosity (inbreeding). The results indicate that the percentage of variance of pedigree-based coancestry estimates explained by genomic coancestry estimates was higher than that for inbreeding. Estimates of effective population size obtained from genome-wide and pedigree information were consistent and ranged from about 66 to 79. These low values emphasize the need of controlling the rate of increase of coancestry and inbreeding in Holstein selection programmes.
尽管荷斯坦奶牛品种的个体数量众多,但其有效种群大小的估计值通常较低。有效种群大小与共同祖先系数和近亲繁殖率呈负相关,由于进行了高强度且准确的选择,这些系数一直很高。传统上,共同祖先系数和近亲繁殖系数是根据系谱数据计算得出的。然而,全基因组单核苷酸多态性的发展增加了人们从分子数据计算这些系数的兴趣,以便提高其准确性。在本研究中,我们获得了西班牙荷斯坦奶牛群体共同祖先系数、近亲繁殖系数和有效种群大小的基因组估计值,然后将其与基于系谱的估计值进行比较。共有11,135头使用Illumina BovineSNP50 BeadChip进行基因分型的动物可用于本研究。应用筛选标准后,最终的基因组数据集包括36,693个常染色体单核苷酸多态性和10,569头动物。这些基因分型动物的系谱数据包括31,203头动物。为了使各动物的系谱信息量均匀化,这些个体仅代表最近五代。共同祖先系数和近亲繁殖系数的基因组估计值是通过同源片段(共同祖先系数)或纯合子连续片段(近亲繁殖系数)获得的。结果表明,基因组共同祖先系数估计值对基于系谱的共同祖先系数估计值方差的解释百分比高于对近亲繁殖系数的解释百分比。从全基因组和系谱信息获得的有效种群大小估计值是一致的,范围约为66至79。这些较低的值强调了在荷斯坦奶牛选择计划中控制共同祖先系数和近亲繁殖率增长的必要性。