Sborgi Lorenzo, Verma Abhinav, Piana Stefano, Lindorff-Larsen Kresten, Cerminara Michele, Santiveri Clara M, Shaw David E, de Alba Eva, Muñoz Victor
†National Biotechnology Center, CSIC, Madrid 28049, Spain.
‡D. E. Shaw Research, New York, New York 10036, United States.
J Am Chem Soc. 2015 May 27;137(20):6506-16. doi: 10.1021/jacs.5b02324. Epub 2015 May 12.
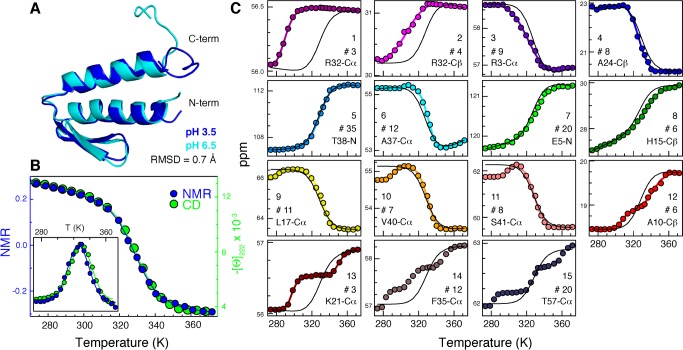
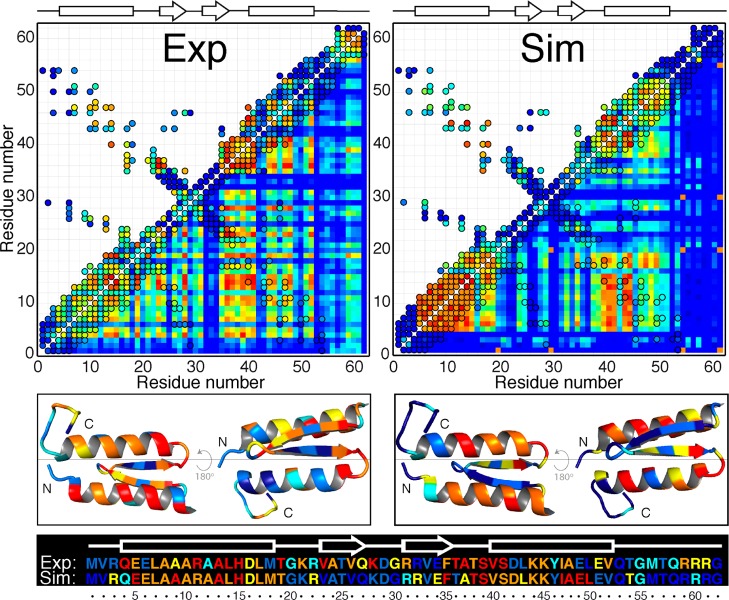
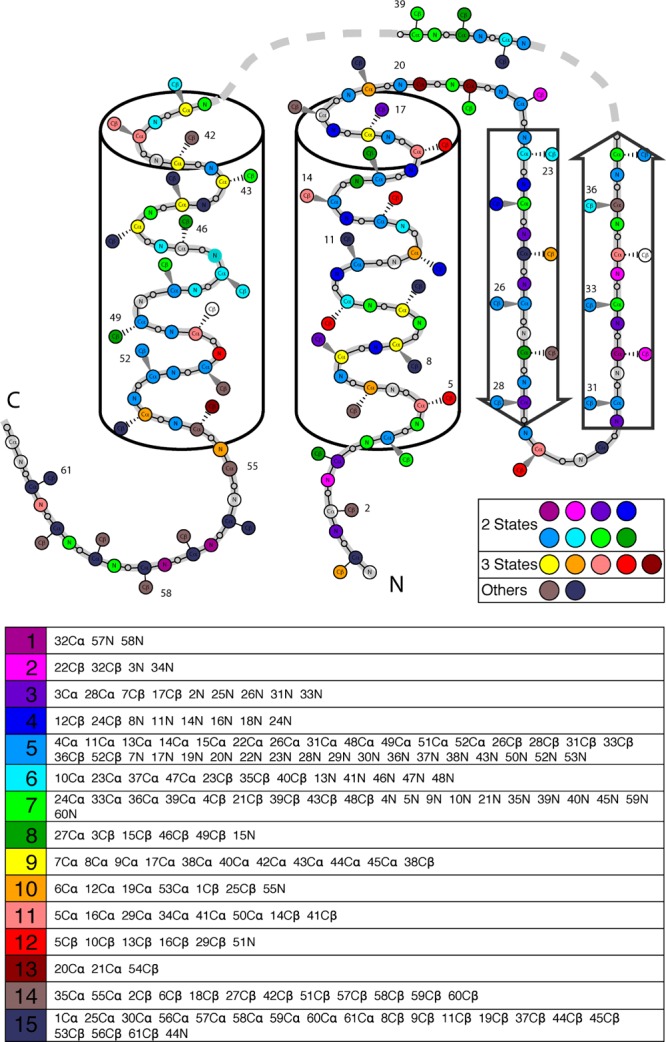
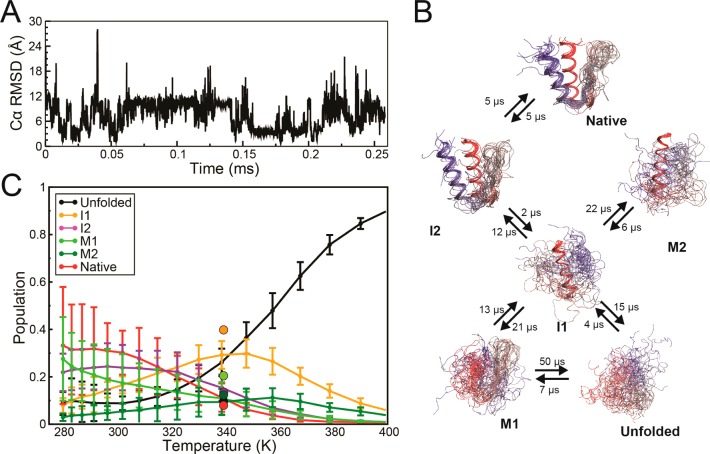
The integration of atomic-resolution experimental and computational methods offers the potential for elucidating key aspects of protein folding that are not revealed by either approach alone. Here, we combine equilibrium NMR measurements of thermal unfolding and long molecular dynamics simulations to investigate the folding of gpW, a protein with two-state-like, fast folding dynamics and cooperative equilibrium unfolding behavior. Experiments and simulations expose a remarkably complex pattern of structural changes that occur at the atomic level and from which the detailed network of residue-residue couplings associated with cooperative folding emerges. Such thermodynamic residue-residue couplings appear to be linked to the order of mechanistically significant events that take place during the folding process. Our results on gpW indicate that the methods employed in this study are likely to prove broadly applicable to the fine analysis of folding mechanisms in fast folding proteins.
原子分辨率实验方法与计算方法的结合,为阐明蛋白质折叠的关键方面提供了可能,而这些方面是单独使用任何一种方法都无法揭示的。在这里,我们结合了热展开的平衡核磁共振测量和长时间分子动力学模拟,来研究gpW的折叠,gpW是一种具有类似两态、快速折叠动力学和协同平衡展开行为的蛋白质。实验和模拟揭示了在原子水平上发生的极其复杂的结构变化模式,与之相关的协同折叠的详细残基-残基耦合网络也从中显现出来。这种热力学残基-残基耦合似乎与折叠过程中发生的具有重要机制意义的事件顺序有关。我们对gpW的研究结果表明,本研究中采用的方法可能被广泛应用于快速折叠蛋白质折叠机制的精细分析。