Hirbe Angela C, Dahiya Sonika, Miller Christopher A, Li Tiandao, Fulton Robert S, Zhang Xiaochun, McDonald Sandra, DeSchryver Katherine, Duncavage Eric J, Walrath Jessica, Reilly Karlyne M, Abel Haley J, Pekmezci Melike, Perry Arie, Ley Timothy J, Gutmann David H
Division of Medical Oncology, Department of Medicine, Washington University School of Medicine, St. Louis, Missouri.
Department of Pathology and Immunology, Washington University School of Medicine, St. Louis, Missouri.
Clin Cancer Res. 2015 Sep 15;21(18):4201-11. doi: 10.1158/1078-0432.CCR-14-3049. Epub 2015 Apr 29.
Malignant peripheral nerve sheath tumors (MPNST) occur at increased frequency in individuals with neurofibromatosis type 1 (NF1), where they likely arise from benign plexiform neurofibroma precursors. While previous studies have used a variety of discovery approaches to discover genes associated with MPNST pathogenesis, it is currently unclear what molecular events are associated with the evolution of MPNST from plexiform neurofibroma.
Whole-exome sequencing was performed on biopsy materials representing plexiform neurofibroma (n = 3), MPNST, and metastasis from a single individual with NF1 over a 14-year period. Additional validation cases were used to assess candidate genes involved in malignant progression, while a murine MPNST model was used for functional analysis.
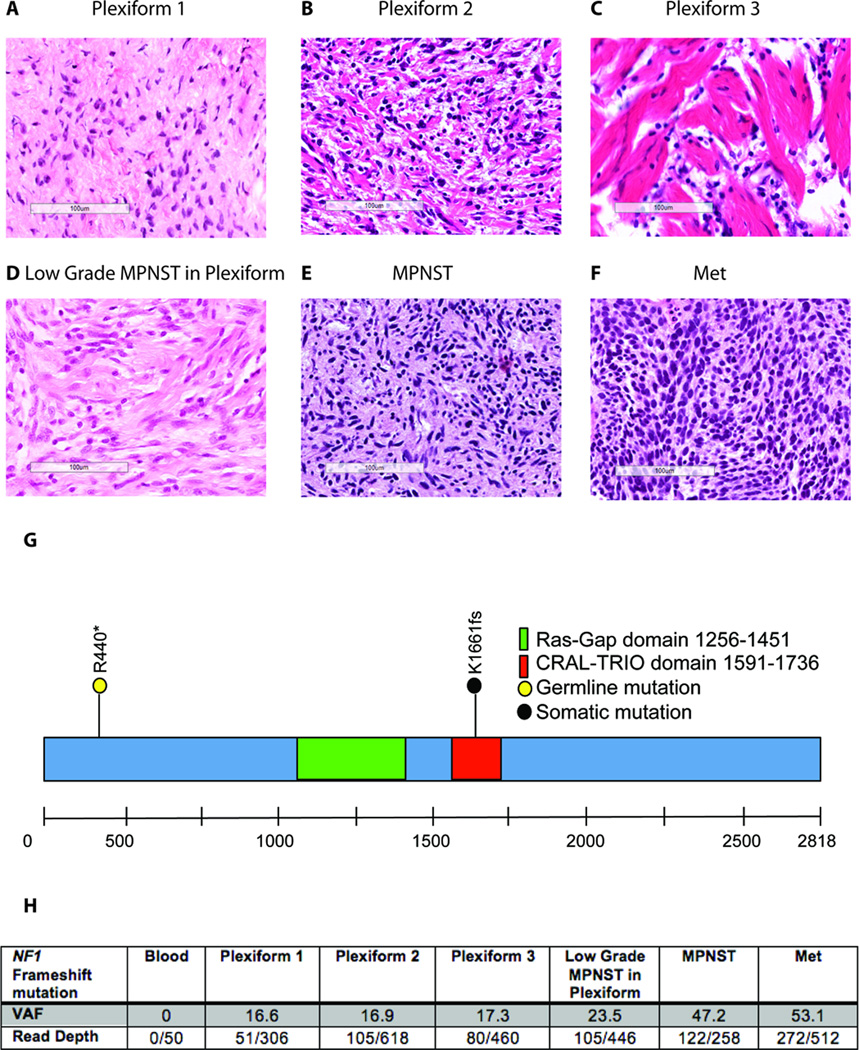
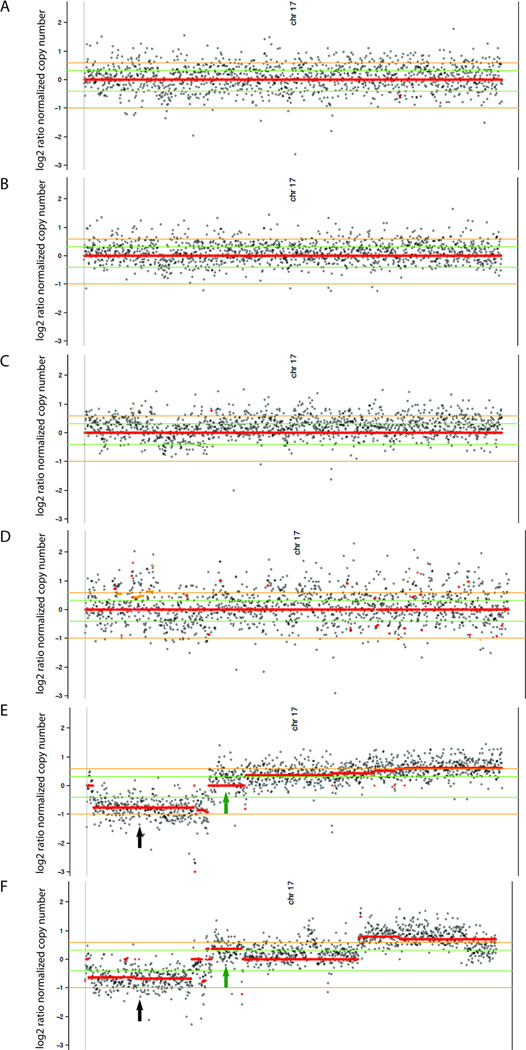
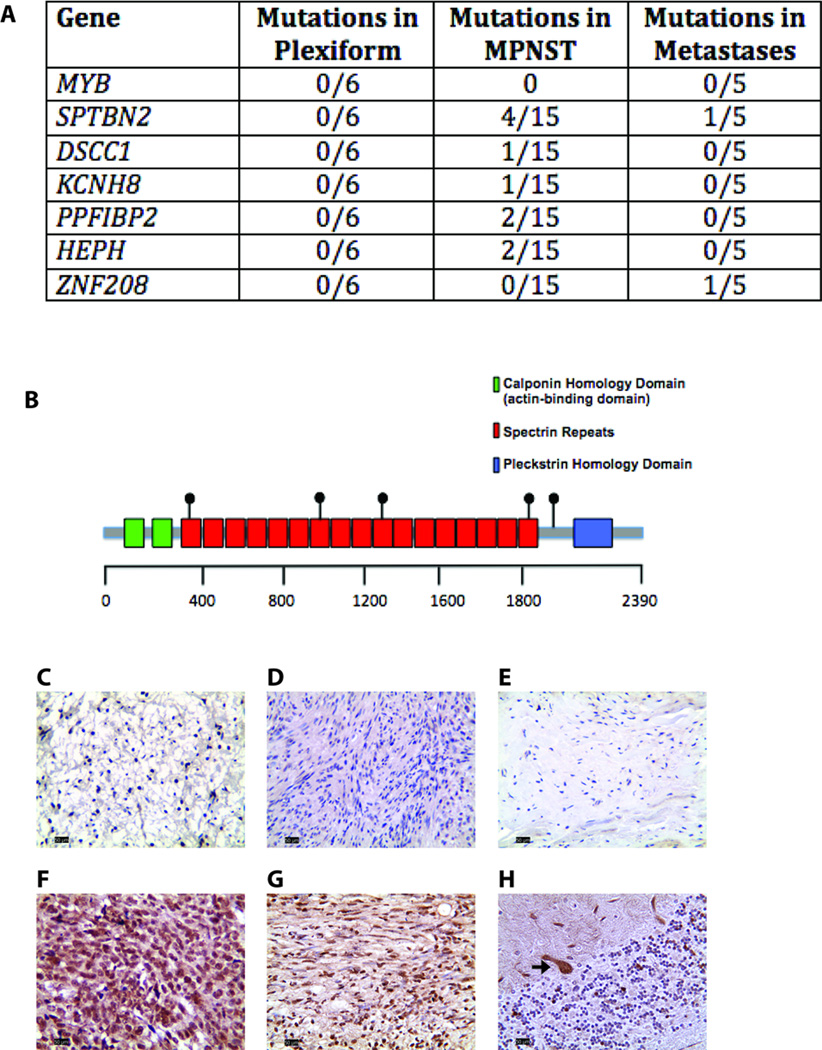
There was an increasing proportion of cells with a somatic NF1 gene mutation as the tumors progressed from benign to malignant, suggesting a clonal process in MPNST development. Copy number variations, including loss of one copy of the TP53 gene, were identified in the primary tumor and the metastatic lesion, but not in benign precursor lesions. A limited number of genes with nonsynonymous somatic mutations (βIII-spectrin and ZNF208) were discovered, several of which were validated in additional primary and metastatic MPNST samples. Finally, increased βIII-spectrin expression was observed in the majority of MPNSTs, and shRNA-mediated knockdown reduced murine MPNST growth in vivo.
Collectively, the ability to track the molecular evolution of MPNST in a single individual with NF1 offers new insights into the sequence of genetic events important for disease pathogenesis and progression for future mechanistic study.
恶性外周神经鞘瘤(MPNST)在1型神经纤维瘤病(NF1)患者中发病率增加,它们可能起源于良性丛状神经纤维瘤前体。虽然先前的研究使用了多种发现方法来发现与MPNST发病机制相关的基因,但目前尚不清楚哪些分子事件与丛状神经纤维瘤向MPNST的演变有关。
对来自一名NF1患者在14年期间的丛状神经纤维瘤(n = 3)、MPNST及转移灶的活检材料进行全外显子组测序。使用额外的验证病例来评估参与恶性进展的候选基因,同时使用小鼠MPNST模型进行功能分析。
随着肿瘤从良性发展为恶性,体细胞NF1基因突变的细胞比例增加,提示MPNST发生过程存在克隆过程。在原发性肿瘤和转移灶中鉴定出拷贝数变异,包括TP53基因一个拷贝的缺失,但在良性前体病变中未发现。发现了少数具有非同义体细胞突变的基因(βIII-血影蛋白和ZNF208),其中一些在额外的原发性和转移性MPNST样本中得到验证。最后,在大多数MPNST中观察到βIII-血影蛋白表达增加,并且shRNA介导的敲低降低了小鼠MPNST在体内的生长。
总体而言,追踪一名NF1患者MPNST分子演变的能力为疾病发病机制和进展中重要的遗传事件序列提供了新见解,以供未来进行机制研究。