Popic Victoria, Salari Raheleh, Hajirasouliha Iman, Kashef-Haghighi Dorna, West Robert B, Batzoglou Serafim
Department of Computer Science, Stanford University, Stanford, CA, USA.
Department of Pathology, Stanford University School of Medicine, Stanford, CA, USA.
Genome Biol. 2015 May 6;16(1):91. doi: 10.1186/s13059-015-0647-8.
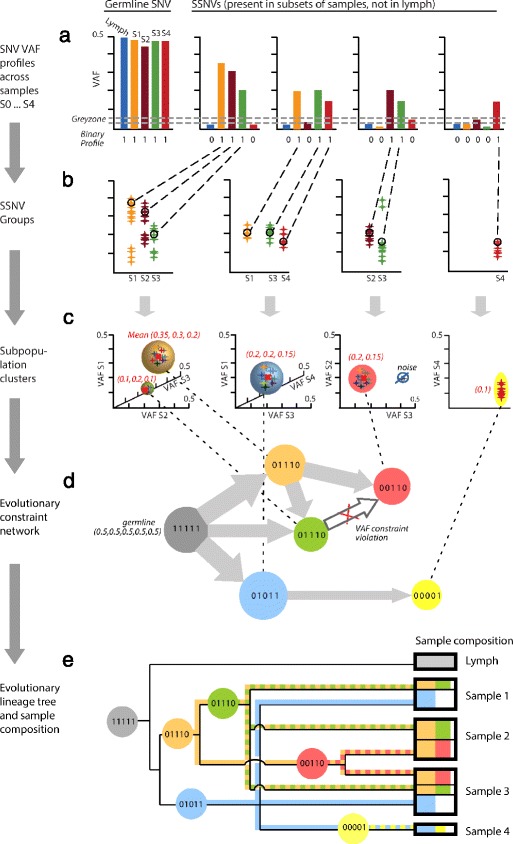
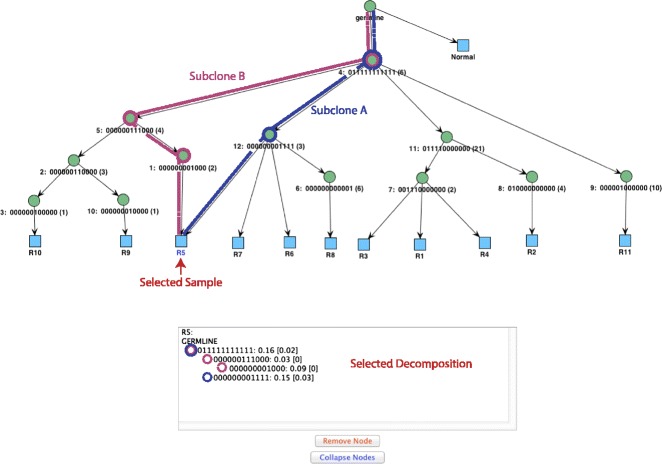
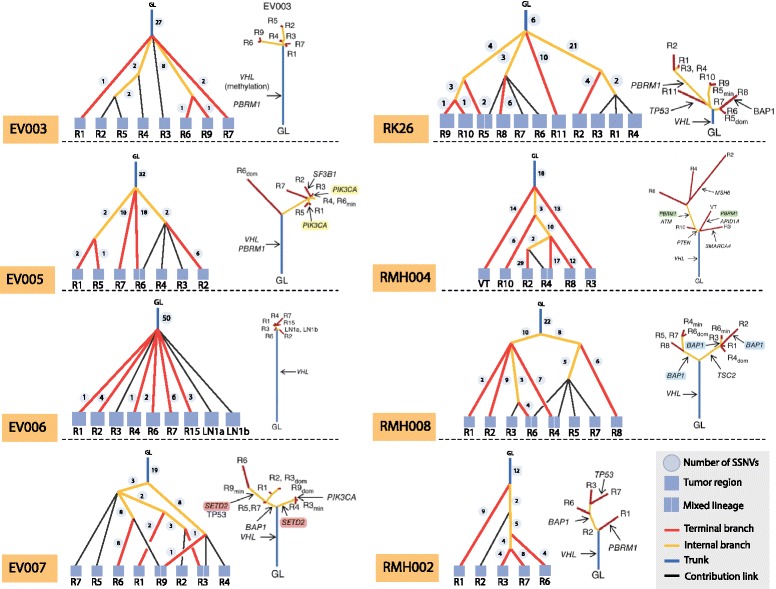
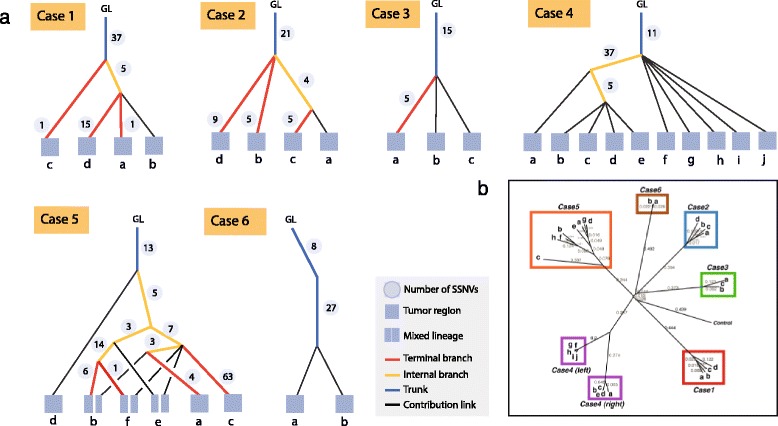
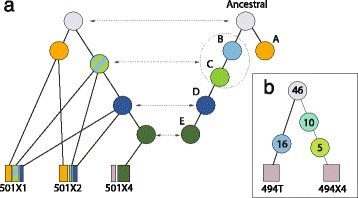
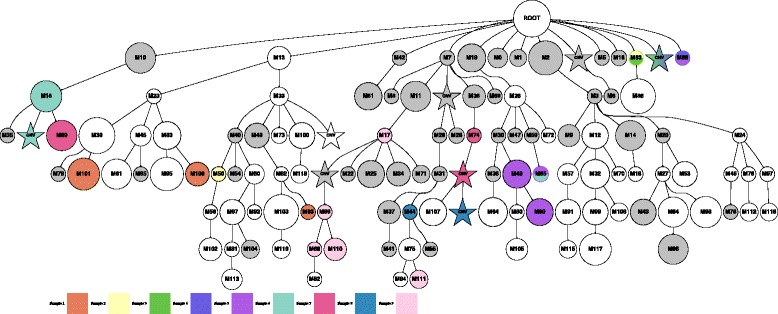
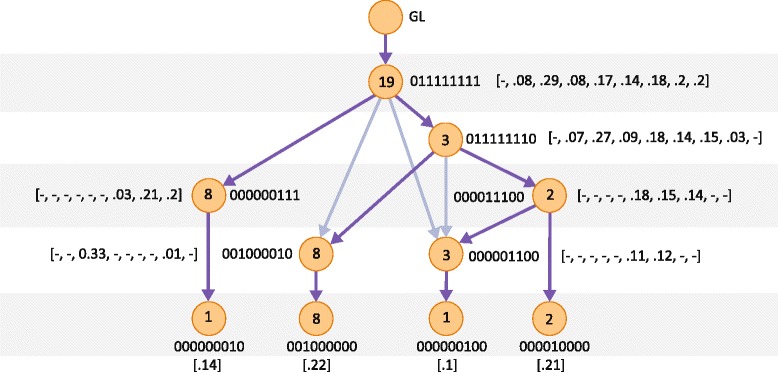
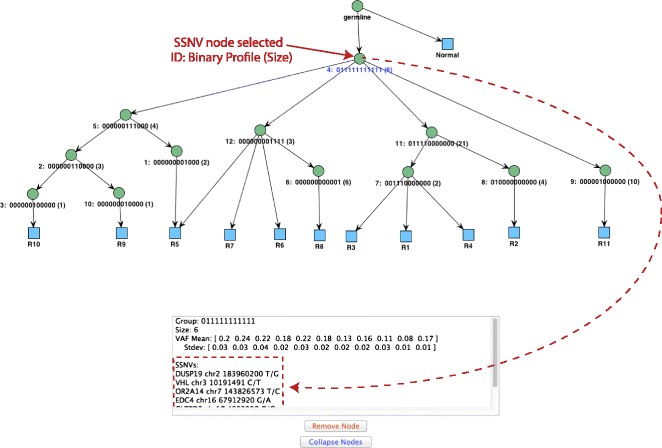
Somatic variants can be used as lineage markers for the phylogenetic reconstruction of cancer evolution. Since somatic phylogenetics is complicated by sample heterogeneity, novel specialized tree-building methods are required for cancer phylogeny reconstruction. We present LICHeE (Lineage Inference for Cancer Heterogeneity and Evolution), a novel method that automates the phylogenetic inference of cancer progression from multiple somatic samples. LICHeE uses variant allele frequencies of somatic single nucleotide variants obtained by deep sequencing to reconstruct multi-sample cell lineage trees and infer the subclonal composition of the samples. LICHeE is open source and available at http://viq854.github.io/lichee .
体细胞变异可用作癌症进化系统发育重建的谱系标记。由于体细胞系统发育因样本异质性而变得复杂,因此癌症系统发育重建需要新的专门建树方法。我们提出了LICHeE(癌症异质性与进化谱系推断),这是一种从多个体细胞样本自动推断癌症进展系统发育的新方法。LICHeE利用通过深度测序获得的体细胞单核苷酸变异的变异等位基因频率来重建多样本细胞谱系树,并推断样本的亚克隆组成。LICHeE是开源的,可在http://viq854.github.io/lichee获取。