McCourt Jackie L, Rhett Katrina K, Jaeger Michele A, Belanto Joseph J, Talsness Dana M, Ervasti James M
Department of Biochemistry, Molecular Biology, and Biophysics, University of Minnesota - Twin Cities, Minneapolis, MN 55455 USA.
Skelet Muscle. 2015 Apr 28;5:13. doi: 10.1186/s13395-015-0040-z. eCollection 2015.
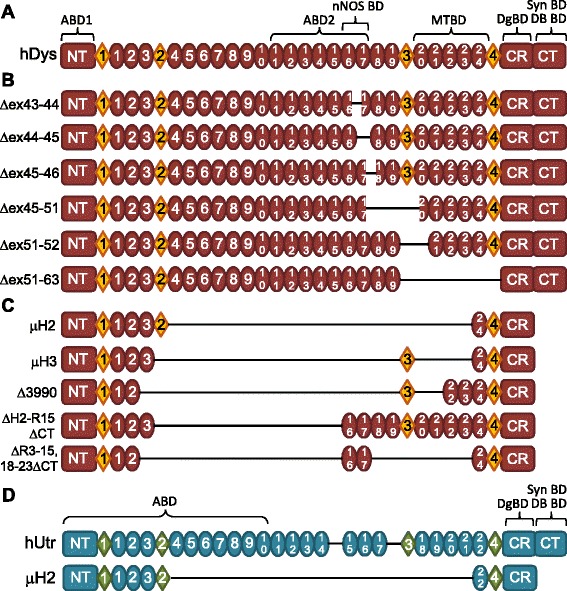
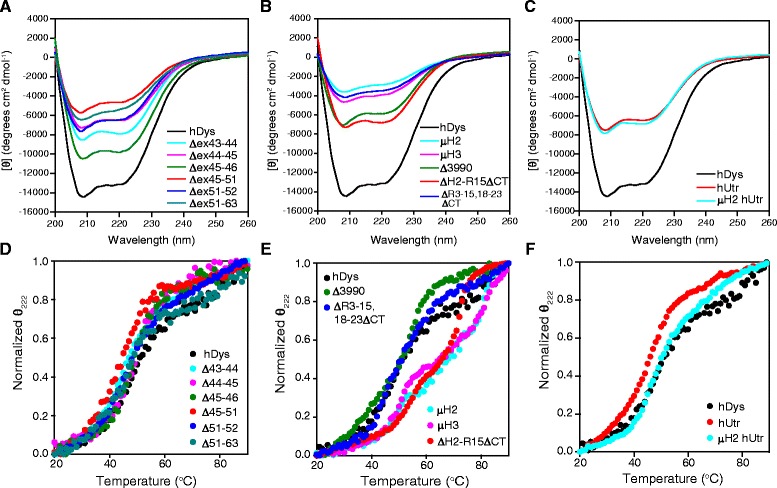
The X-linked recessive disease Duchenne muscular dystrophy (DMD) is caused by mutations in the gene encoding the protein dystrophin. Despite its large size, dystrophin is a highly stable protein, demonstrating cooperative unfolding during thermal denaturation as monitored by circular dichroism spectroscopy. In contrast, internal sequence deletions have been associated with a loss of the cooperative unfolding and cause in vitro protein aggregation. Several emerging therapy options for DMD utilize internally deleted micro-dystrophins and multi-exon-skipped dystrophins that produce partially functional proteins, but the stability of such internally truncated proteins has not been investigated.
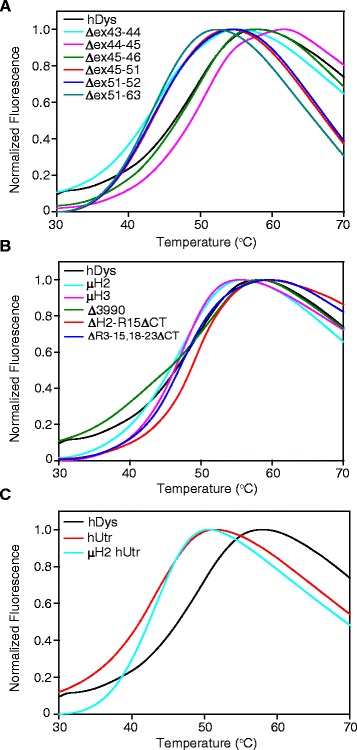
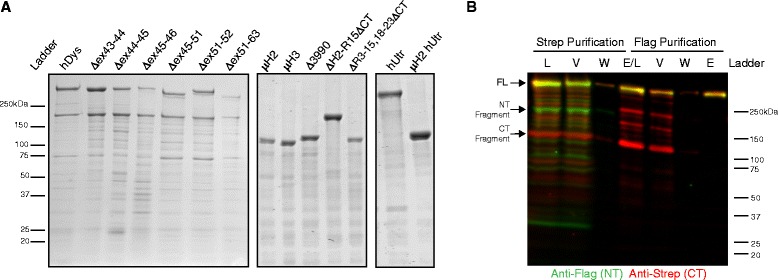
In this study, we analyzed the in vitro stability of human dystrophin constructs skipped around exon 45 or exon 51, several dystrophin gene therapy constructs, as well as human full-length and micro-utrophin. Constructs were expressed in insect cells using the baculovirus system, purified by affinity chromatography, and analyzed by high-speed sedimentation, circular dichroism spectroscopy, and differential scanning fluorimetry.
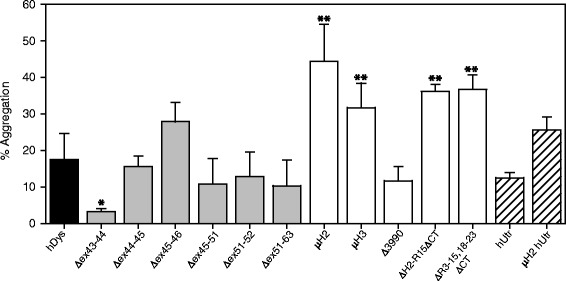
Our results reveal that not all gene therapy constructs display stabilities consistent with full-length human dystrophin. However, all dystrophins skipped in-frame around exon 45 or exon 51 show stability profiles congruent with intact human dystrophin. Similar to previous studies of mouse proteins, full-length human utrophin also displays stability similar to human dystrophin and does not appear to be affected by a large internal deletion.
Our results suggest that the in vitro stability of human dystrophin is less sensitive to smaller deletions at natural exon boundaries than larger, more complex deletions present in some gene therapy constructs.
X连锁隐性疾病杜氏肌营养不良症(DMD)由编码抗肌萎缩蛋白的基因突变引起。尽管抗肌萎缩蛋白体积庞大,但它是一种高度稳定的蛋白质,在圆二色光谱监测下,热变性过程中呈现协同解折叠。相比之下,内部序列缺失与协同解折叠的丧失有关,并导致体外蛋白质聚集。几种新兴的DMD治疗方案利用内部缺失的微型抗肌萎缩蛋白和多外显子跳跃抗肌萎缩蛋白,这些蛋白可产生部分功能性蛋白质,但此类内部截短蛋白的稳定性尚未得到研究。
在本研究中,我们分析了跨越外显子45或外显子51跳跃的人抗肌萎缩蛋白构建体、几种抗肌萎缩蛋白基因治疗构建体以及人全长和微型肌养蛋白的体外稳定性。构建体在昆虫细胞中利用杆状病毒系统表达,通过亲和层析纯化,并通过高速沉降、圆二色光谱和差示扫描荧光法进行分析。
我们的结果表明,并非所有基因治疗构建体都表现出与全长人抗肌萎缩蛋白一致的稳定性。然而;所有在框架内跨越外显子45或外显子51跳跃的抗肌萎缩蛋白都显示出与完整人抗肌萎缩蛋白一致的稳定性特征。与先前对小鼠蛋白的研究类似,全长人肌养蛋白也表现出与人抗肌萎缩蛋白相似的稳定性,并且似乎不受大的内部缺失的影响。
我们的结果表明,人抗肌萎缩蛋白的体外稳定性对天然外显子边界处较小的缺失不如某些基因治疗构建体中存在的较大、更复杂的缺失敏感。