Gorfine Malka, Goldstein Boaz, Fishman Alla, Heller Ruth, Heller Yair, Lamm Ayelet T
Faculty of Industrial Engineering and Management, Technion- Israel Institute of Technology, Technion City, Haifa 3200003, Israel.
Faculty of Biology, Technion- Israel Institute of Technology, Technion City, Haifa 3200003, Israel.
PLoS One. 2015 May 12;10(5):e0126544. doi: 10.1371/journal.pone.0126544. eCollection 2015.
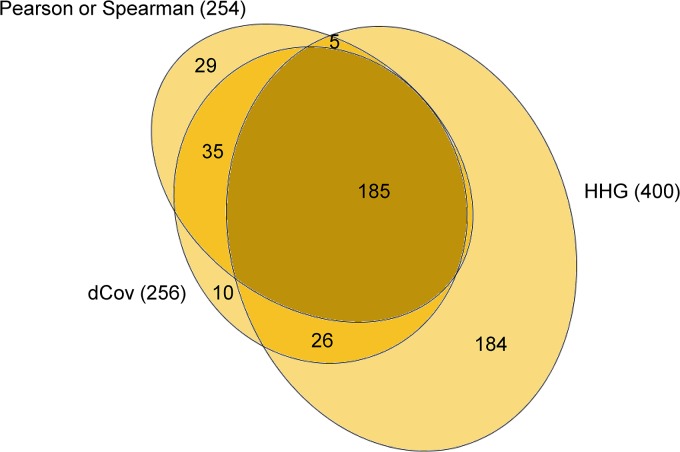
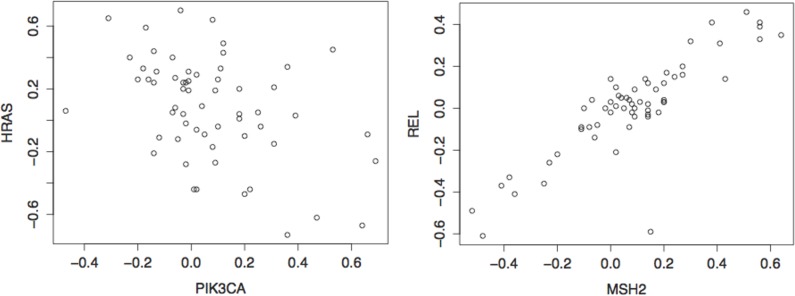
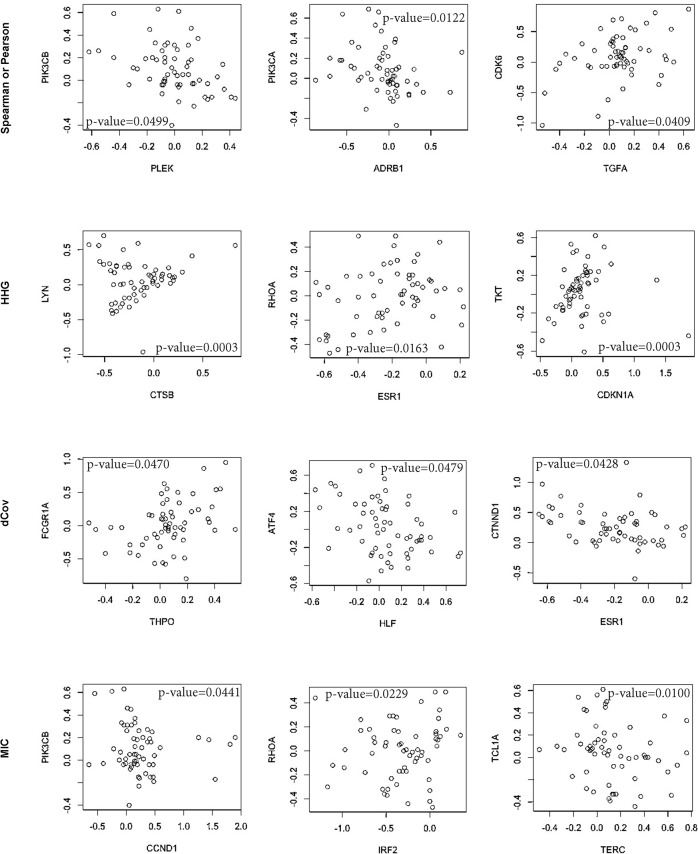
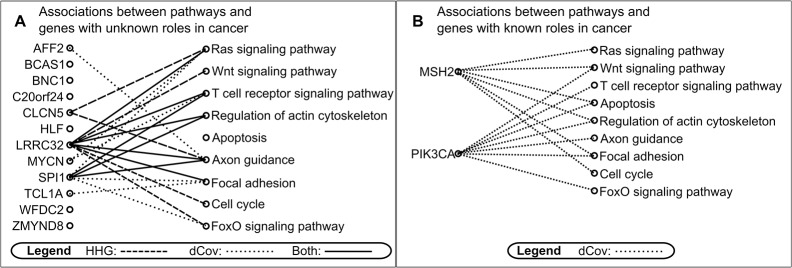
Copy number variation (CNV) plays a role in pathogenesis of many human diseases, especially cancer. Several whole genome CNV association studies have been performed for the purpose of identifying cancer associated CNVs. Here we undertook a novel approach to whole genome CNV analysis, with the goal being identification of associations between CNV of different genes (CNV-CNV) across 60 human cancer cell lines. We hypothesize that these associations point to the roles of the associated genes in cancer, and can be indicators of their position in gene networks of cancer-driving processes. Recent studies show that gene associations are often non-linear and non-monotone. In order to obtain a more complete picture of all CNV associations, we performed omnibus univariate analysis by utilizing dCov, MIC, and HHG association tests, which are capable of detecting any type of association, including non-monotone relationships. For comparison we used Spearman and Pearson association tests, which detect only linear or monotone relationships. Application of dCov, MIC and HHG tests resulted in identification of twice as many associations compared to those found by Spearman and Pearson alone. Interestingly, most of the new associations were detected by the HHG test. Next, we utilized dCov's and HHG's ability to perform multivariate analysis. We tested for association between genes of unknown function and known cancer-related pathways. Our results indicate that multivariate analysis is much more effective than univariate analysis for the purpose of ascribing biological roles to genes of unknown function. We conclude that a combination of multivariate and univariate omnibus association tests can reveal significant information about gene networks of disease-driving processes. These methods can be applied to any large gene or pathway dataset, allowing more comprehensive analysis of biological processes.
拷贝数变异(CNV)在许多人类疾病尤其是癌症的发病机制中发挥作用。为了识别与癌症相关的CNV,已经开展了多项全基因组CNV关联研究。在此,我们采用了一种全新的全基因组CNV分析方法,目标是识别60种人类癌细胞系中不同基因的CNV之间的关联(CNV-CNV)。我们假设这些关联指向相关基因在癌症中的作用,并且可以作为它们在癌症驱动过程基因网络中位置的指标。最近的研究表明,基因关联通常是非线性和非单调的。为了更全面地了解所有CNV关联,我们利用能够检测任何类型关联(包括非单调关系)的dCov、MIC和HHG关联检验进行了综合单变量分析。为了进行比较,我们使用了仅能检测线性或单调关系的Spearman和Pearson关联检验。与仅使用Spearman和Pearson检验相比,应用dCov、MIC和HHG检验发现的关联数量多出一倍。有趣的是,大多数新关联是通过HHG检验检测到的。接下来,我们利用dCov和HHG进行多变量分析的能力。我们测试了未知功能基因与已知癌症相关通路之间的关联。我们的结果表明,就为未知功能基因赋予生物学作用而言,多变量分析比单变量分析有效得多。我们得出结论,多变量和单变量综合关联检验相结合可以揭示有关疾病驱动过程基因网络的重要信息。这些方法可以应用于任何大型基因或通路数据集,从而对生物学过程进行更全面的分析。