Leong Ivone U S, Stuckey Alexander, Lai Daniel, Skinner Jonathan R, Love Donald R
Diagnostic Genetics, LabPlus, Auckland City Hospital, Auckland, New Zealand.
Bioinformatics Institute, University of Auckland, Auckland, New Zealand.
BMC Med Genet. 2015 May 13;16:34. doi: 10.1186/s12881-015-0176-z.
Long QT syndrome (LQTS) is an autosomal dominant condition predisposing to sudden death from malignant arrhythmia. Genetic testing identifies many missense single nucleotide variants of uncertain pathogenicity. Establishing genetic pathogenicity is an essential prerequisite to family cascade screening. Many laboratories use in silico prediction tools, either alone or in combination, or metaservers, in order to predict pathogenicity; however, their accuracy in the context of LQTS is unknown. We evaluated the accuracy of five in silico programs and two metaservers in the analysis of LQTS 1-3 gene variants.
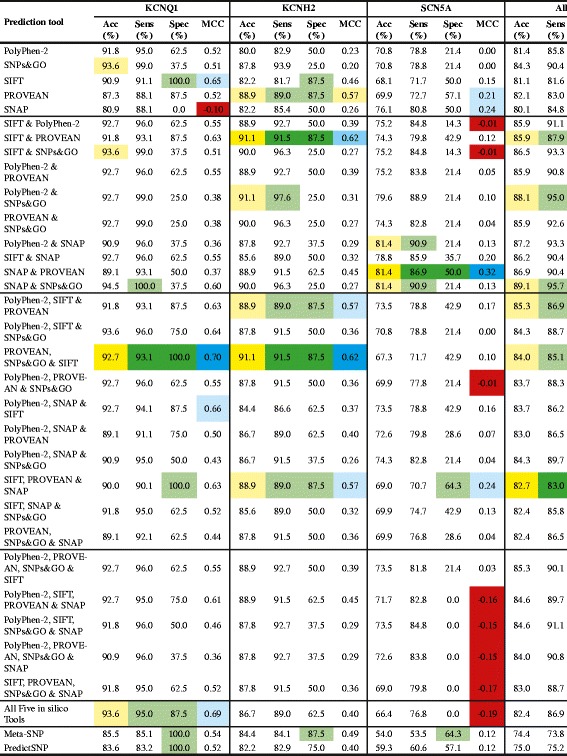
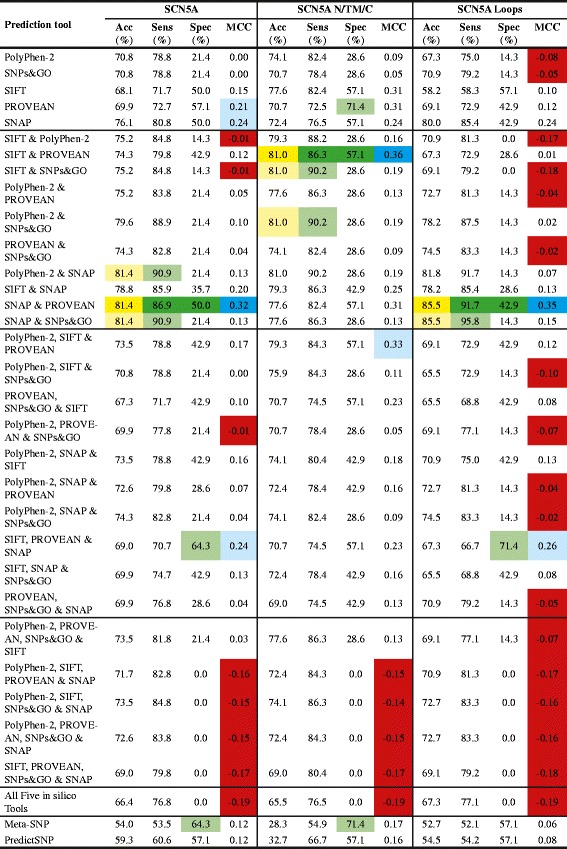
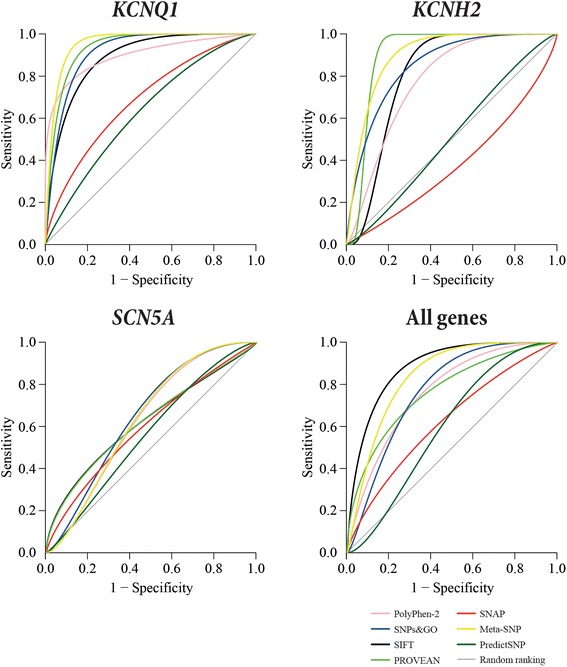
The in silico tools SIFT, PolyPhen-2, PROVEAN, SNPs&GO and SNAP, either alone or in all possible combinations, and the metaservers Meta-SNP and PredictSNP, were tested on 312 KCNQ1, KCNH2 and SCN5A gene variants that have previously been characterised by either in vitro or co-segregation studies as either "pathogenic" (283) or "benign" (29). The accuracy, sensitivity, specificity and Matthews Correlation Coefficient (MCC) were calculated to determine the best combination of in silico tools for each LQTS gene, and when all genes are combined.
The best combination of in silico tools for KCNQ1 is PROVEAN, SNPs&GO and SIFT (accuracy 92.7%, sensitivity 93.1%, specificity 100% and MCC 0.70). The best combination of in silico tools for KCNH2 is SIFT and PROVEAN or PROVEAN, SNPs&GO and SIFT. Both combinations have the same scores for accuracy (91.1%), sensitivity (91.5%), specificity (87.5%) and MCC (0.62). In the case of SCN5A, SNAP and PROVEAN provided the best combination (accuracy 81.4%, sensitivity 86.9%, specificity 50.0%, and MCC 0.32). When all three LQT genes are combined, SIFT, PROVEAN and SNAP is the combination with the best performance (accuracy 82.7%, sensitivity 83.0%, specificity 80.0%, and MCC 0.44). Both metaservers performed better than the single in silico tools; however, they did not perform better than the best performing combination of in silico tools.
The combination of in silico tools with the best performance is gene-dependent. The in silico tools reported here may have some value in assessing variants in the KCNQ1 and KCNH2 genes, but caution should be taken when the analysis is applied to SCN5A gene variants.
长QT综合征(LQTS)是一种常染色体显性疾病,易发生恶性心律失常导致猝死。基因检测可识别许多致病性不确定的错义单核苷酸变异。确定基因致病性是家族级联筛查的重要前提。许多实验室单独或联合使用计算机预测工具或元服务器来预测致病性;然而,它们在LQTS背景下的准确性尚不清楚。我们评估了五个计算机程序和两个元服务器在分析LQTS 1 - 3基因变异中的准确性。
对312个先前通过体外或共分离研究被鉴定为“致病性”(283个)或“良性”(29个)的KCNQ1、KCNH2和SCN5A基因变异,分别单独或所有可能组合地测试计算机工具SIFT、PolyPhen - 2、PROVEAN、SNPs&GO和SNAP,以及元服务器Meta - SNP和PredictSNP。计算准确性、敏感性、特异性和马修斯相关系数(MCC),以确定每个LQTS基因以及所有基因组合时计算机工具的最佳组合。
KCNQ1的计算机工具最佳组合是PROVEAN、SNPs&GO和SIFT(准确性92.7%,敏感性93.1%,特异性100%,MCC 0.70)。KCNH2的计算机工具最佳组合是SIFT和PROVEAN或PROVEAN、SNPs&GO和SIFT。两种组合在准确性(91.1%)、敏感性(91.5%)、特异性(87.5%)和MCC(0.62)方面得分相同。对于SCN5A,SNAP和PROVEAN提供了最佳组合(准确性81.4%,敏感性86.9%,特异性50.0%,MCC 0.32)。当三个LQT基因组合在一起时,SIFT、PROVEAN和SNAP是性能最佳的组合(准确性82.7%,敏感性83.0%,特异性80.0%,MCC 0.44)。两个元服务器的表现均优于单个计算机工具;然而,它们的表现不如计算机工具的最佳组合。
性能最佳的计算机工具组合因基因而异。本文报道的计算机工具在评估KCNQ1和KCNH2基因变异时可能有一定价值,但在应用于SCN5A基因变异分析时应谨慎。