Bureau Hailey R, Merz Dale R, Hershkovits Eli, Quirk Stephen, Hernandez Rigoberto
Center for Computational and Molecular Science and Technology, School of Chemistry and Biochemistry, Georgia Institute of Technology, Atlanta, Georgia 30332-0400, United States of America.
Kimberly-Clark Corporation, Atlanta, GA 30076-2199, United States of America.
PLoS One. 2015 May 13;10(5):e0127034. doi: 10.1371/journal.pone.0127034. eCollection 2015.
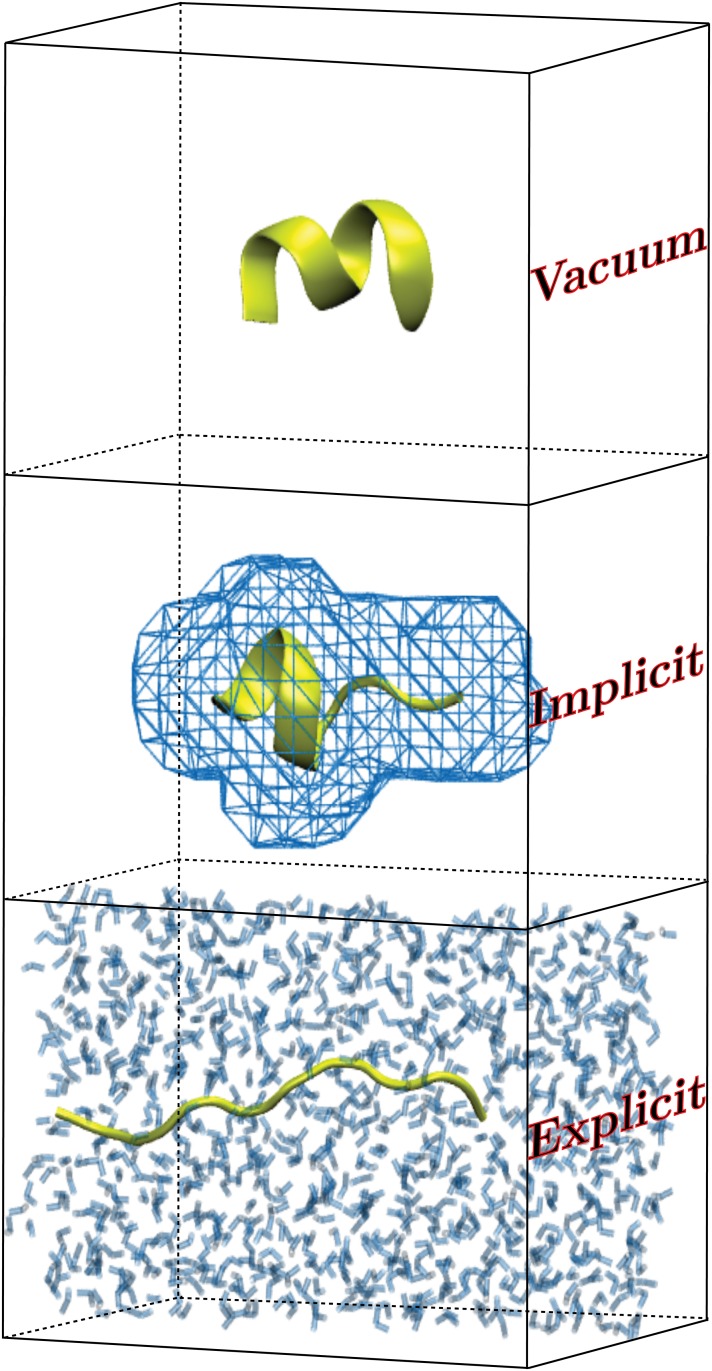
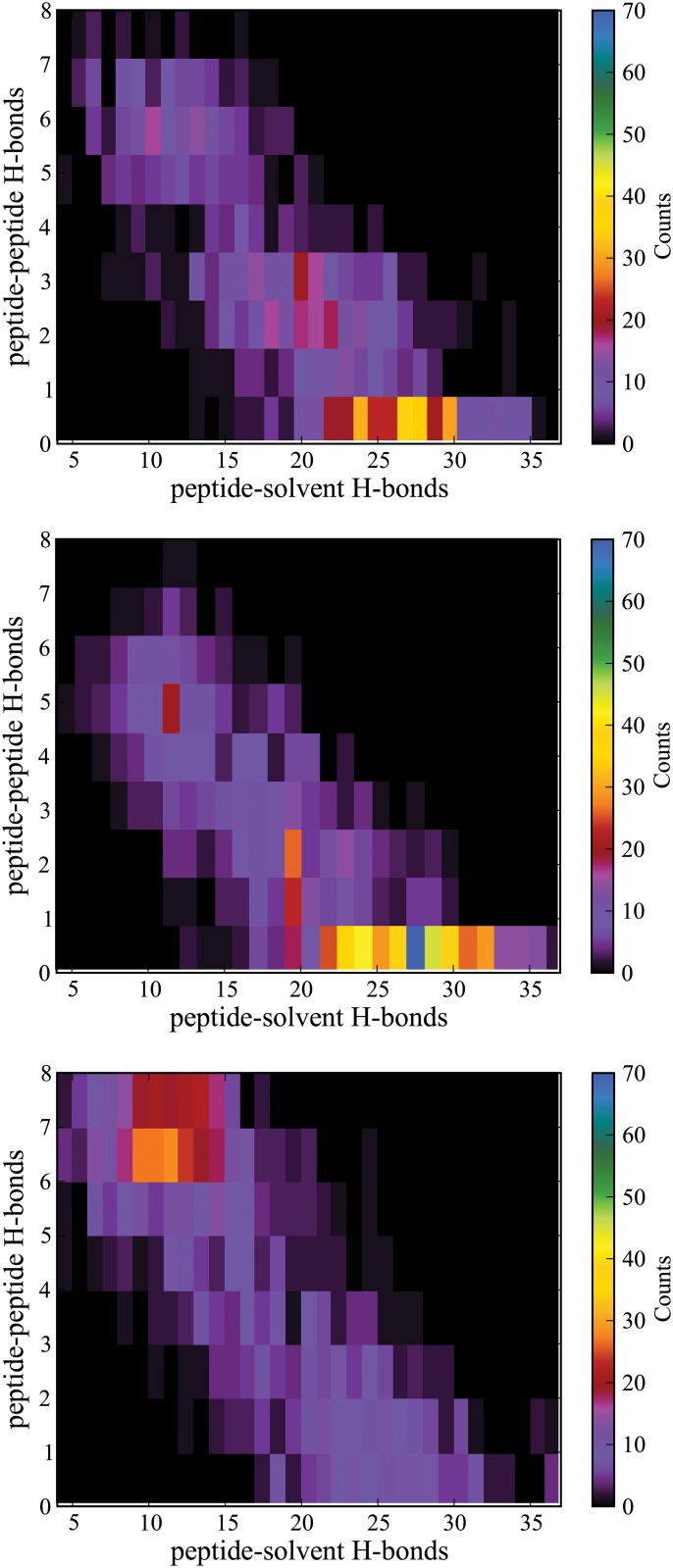
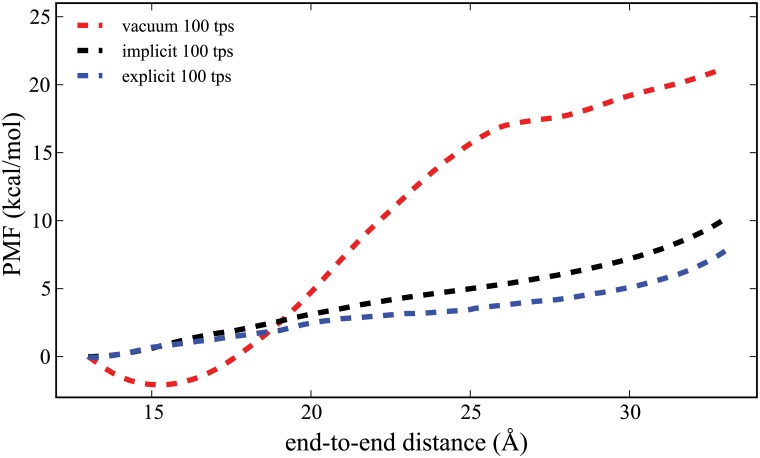
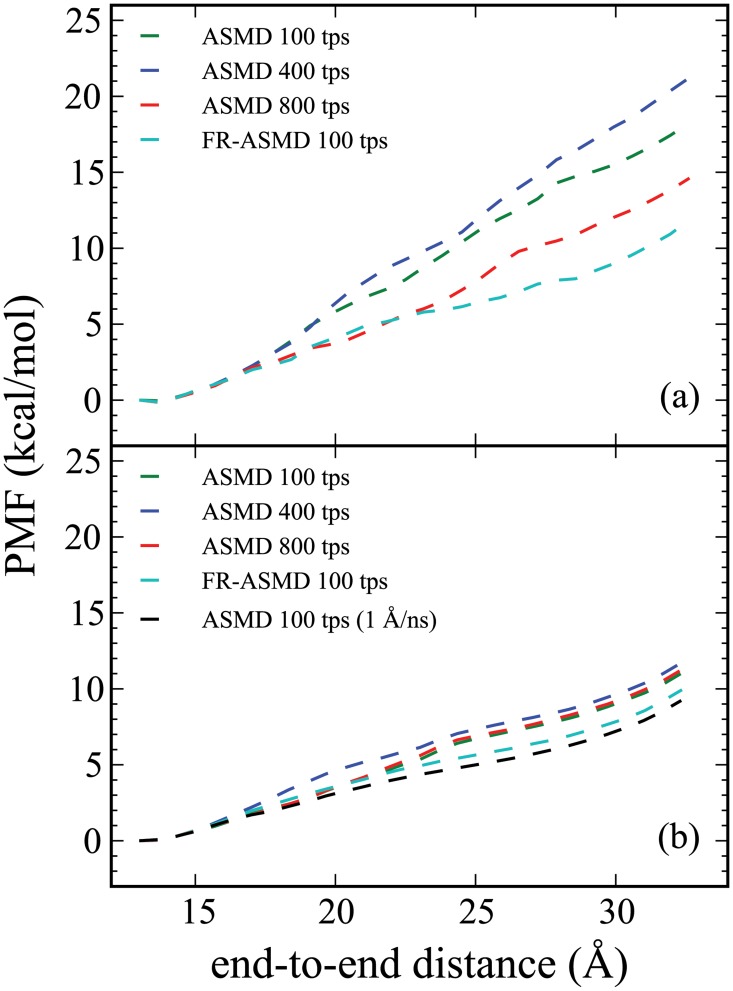
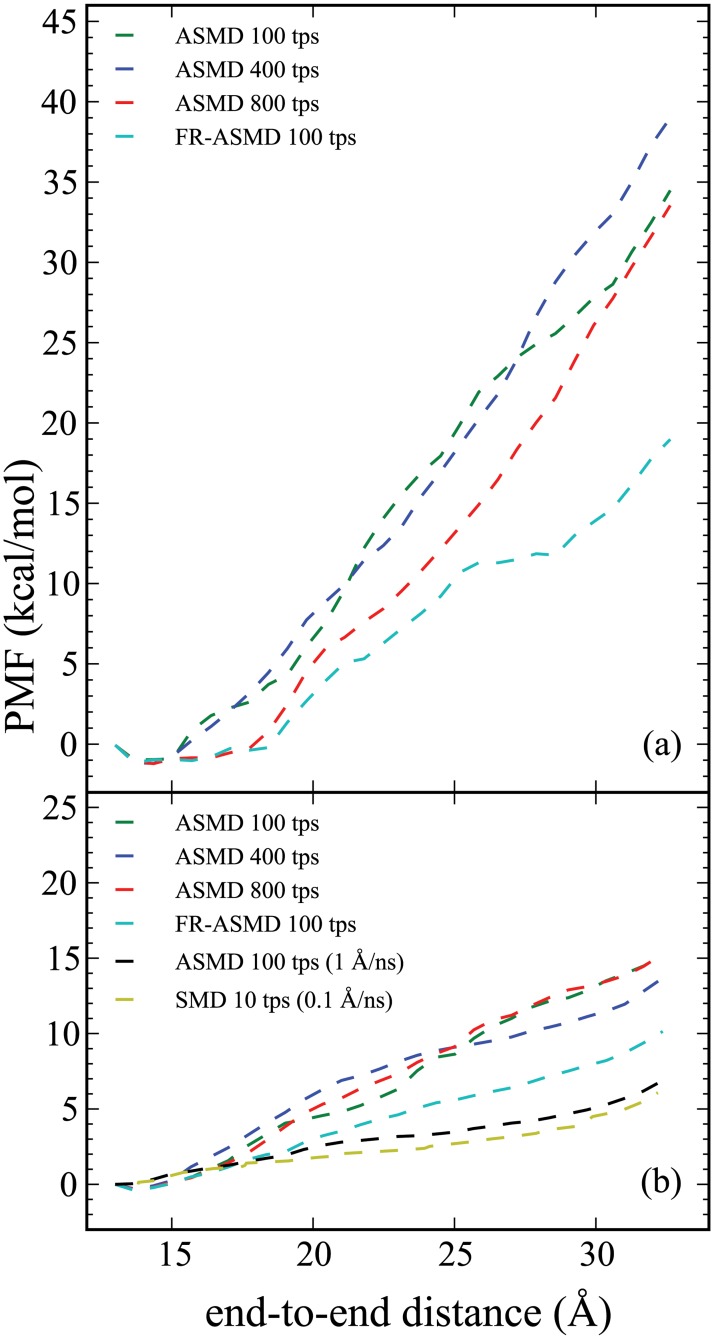
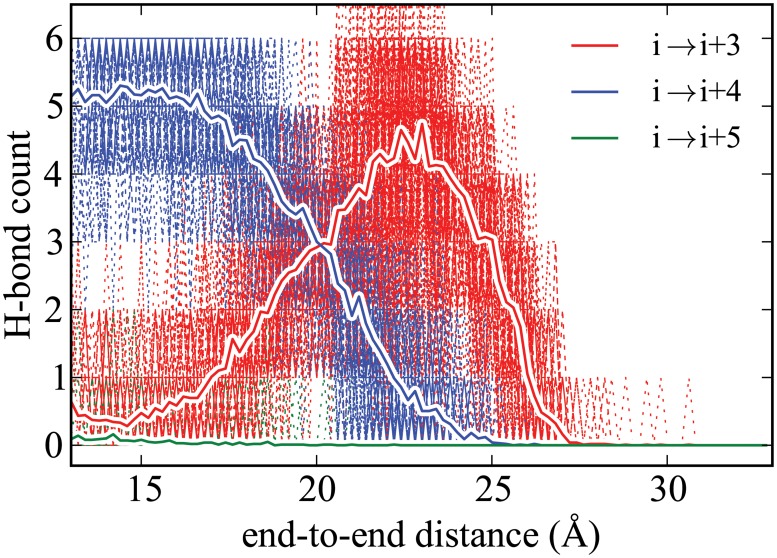
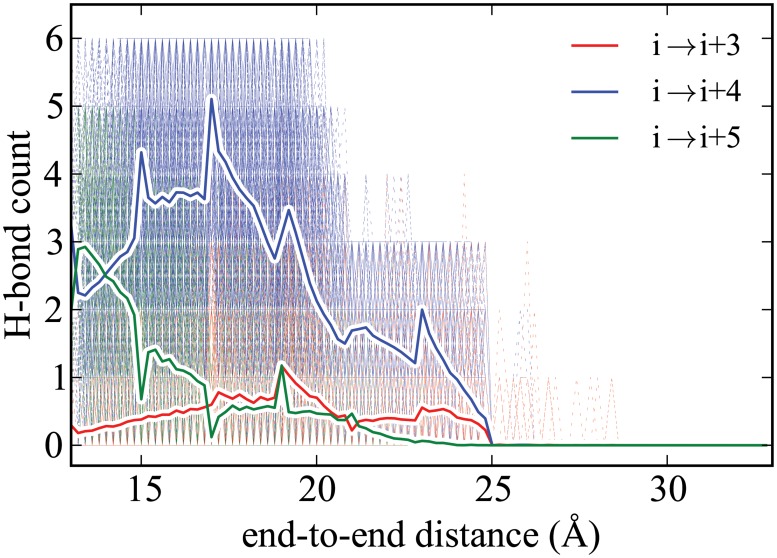
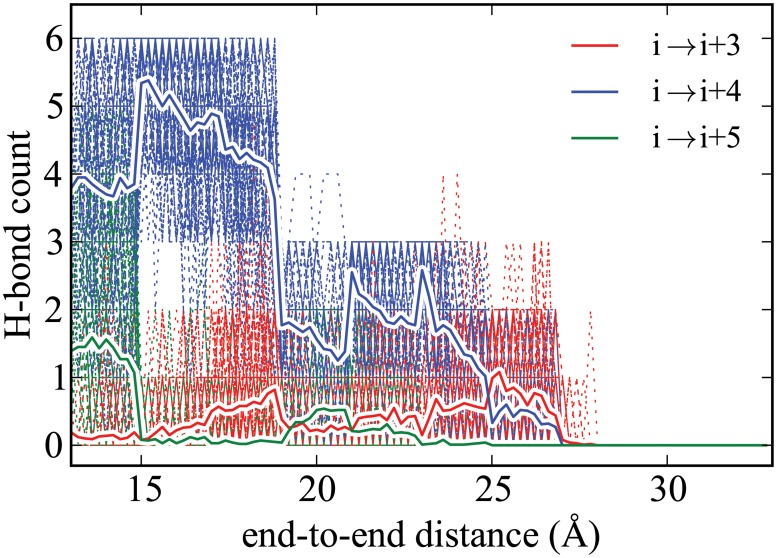
Steered Molecular Dynamics (SMD) has been seen to provide the potential of mean force (PMF) along a peptide unfolding pathway effectively but at significant computational cost, particularly in all-atom solvents. Adaptive steered molecular dynamics (ASMD) has been seen to provide a significant computational advantage by limiting the spread of the trajectories in a staged approach. The contraction of the trajectories at the end of each stage can be performed by taking a structure whose nonequilibrium work is closest to the Jarzynski average (in naive ASMD) or by relaxing the trajectories under a no-work condition (in full-relaxation ASMD--namely, FR-ASMD). Both approaches have been used to determine the energetics and hydrogen-bonding structure along the pathway for unfolding of a benchmark peptide initially constrained as an α-helix in a water environment. The energetics are quite different to those in vacuum, but are found to be similar between implicit and explicit solvents. Surprisingly, the hydrogen-bonding pathways are also similar in the implicit and explicit solvents despite the fact that the solvent contact plays an important role in opening the helix.
已发现,引导分子动力学(SMD)能够有效地沿着肽链解折叠途径提供平均力势(PMF),但计算成本高昂,尤其是在全原子溶剂中。自适应引导分子动力学(ASMD)通过分阶段限制轨迹的扩展,已显示出显著的计算优势。在每个阶段结束时,可以通过采用非平衡功最接近雅尔津斯基平均值的结构(在朴素ASMD中)或在无功条件下使轨迹松弛(在完全松弛ASMD——即FR - ASMD中)来收缩轨迹。这两种方法都已用于确定在水环境中最初被约束为α螺旋的基准肽解折叠途径上的能量学和氢键结构。能量学与真空中的情况有很大不同,但发现在隐式溶剂和显式溶剂之间相似。令人惊讶的是,尽管溶剂接触在打开螺旋中起重要作用,但在隐式溶剂和显式溶剂中氢键途径也相似。