Wang Cong, Hai Yabing, Liu Xiaoqing, Liu Nanfang, Yao Yuhua, He Pingan, Dai Qi
College of Life Sciences, Zhejiang Sci-Tech University, Hangzhou 310018, China.
College of Sciences, Hangzhou Dianzi University, Hangzhou 310018, China.
Comput Math Methods Med. 2015;2015:756345. doi: 10.1155/2015/756345. Epub 2015 Apr 20.
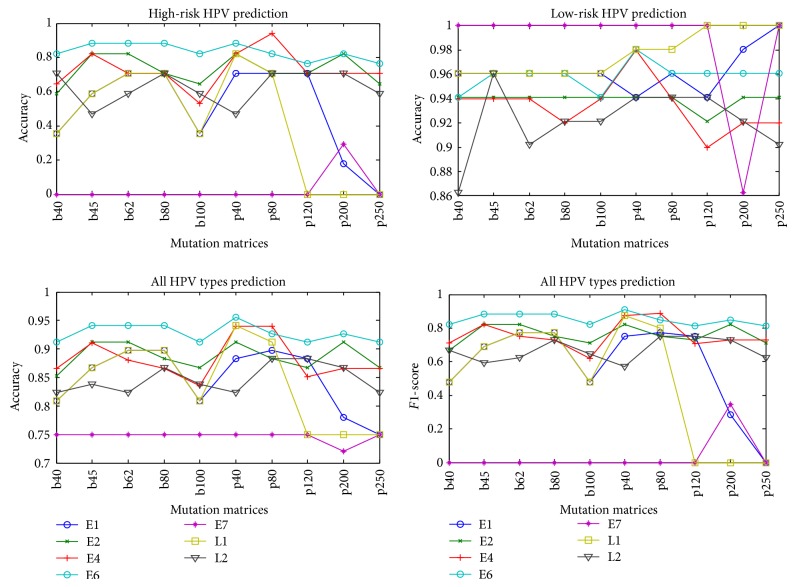
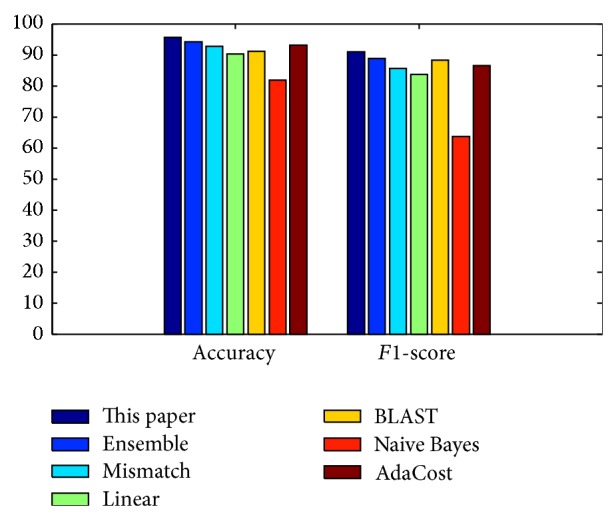
Discrimination of high-risk types of human papillomaviruses plays an important role in the diagnosis and remedy of cervical cancer. Recently, several computational methods have been proposed based on protein sequence-based and structure-based information, but the information of their related proteins has not been used until now. In this paper, we proposed using protein "sequence space" to explore this information and used it to predict high-risk types of HPVs. The proposed method was tested on 68 samples with known HPV types and 4 samples without HPV types and further compared with the available approaches. The results show that the proposed method achieved the best performance among all the evaluated methods with accuracy 95.59% and F1-score 90.91%, which indicates that protein "sequence space" could potentially be used to improve prediction of high-risk types of HPVs.
人类乳头瘤病毒高危类型的鉴别在宫颈癌的诊断和治疗中起着重要作用。最近,已经提出了几种基于蛋白质序列信息和结构信息的计算方法,但直到现在其相关蛋白质的信息尚未得到利用。在本文中,我们提议利用蛋白质“序列空间”来探索这一信息,并将其用于预测高危型人乳头瘤病毒。所提出的方法在68个已知人乳头瘤病毒类型的样本和4个无人乳头瘤病毒类型的样本上进行了测试,并进一步与现有方法进行了比较。结果表明,在所评估的所有方法中,该方法取得了最佳性能,准确率为95.59%,F1分数为90.91%,这表明蛋白质“序列空间”有可能用于改进高危型人乳头瘤病毒的预测。